Human Cytome Project - Building better bridges from molecules to man
![[construction icon]](./images/worker.gif)
Introduction
A human cytome project aims at creating a better understanding of a cellular level of biological complexity in order to allow us to close the gap between (our) molecules and the intrahuman ecosystem. Understanding the (heterogeneous) cellular level of biological organisation and complexity is (almost) within reach of present day science, which makes such a project ambitious but achievable. A human cytome project is about creating a solid translational science, not from bench to bedside, but from molecule to man.
This
document deals with concepts on the exploration of the Human Cytome.
The first part deals with the problems of
analyzing the cytome at multiple levels of biological organization.
The second part deals with the ways of
exploring and analyzing the cytome at the different levels of biological organization
and complexity.
Figure 1. The bridge between the genome and the human biosystem is still not clear. |
The link between genome and the human biosystem
is still not clear (Figure 1). Clinical reality extends beyond
the frontiers of basic science. Outside the boundaries of basic scientific research, such as genomics and proteomics,
significant parts of (biological/clinical) reality remain un-explained for and
not well understood. Research on the multilevel complexity of biological systems has to build the
bridge between basic biological research (genes, proteins) and clinical application. See also: Drug Discovery and Development - Human Cytome Project.
"The accelerating pace of scientific and technological progress which made it possible,
within just a decade, to complete the first full sequencing of the human genome and of a growing number of other living organisms is heralding a new era in
molecular biology and genetics, in particular for human medicine. However, it
is going to take a very large-scale and long-term research effort if the promises
of this 'post-genomic age' are to be realised." (from
The new picture of health).
The purpose of critical discussion is to advance the understanding of the field. While many are spurred to criticize from competitive instincts,
"a discussion which you win but which fails to help ... clarify ... should be regarded as a sheer
loss." (Popper).
We may now be capable to
study a low-level layer of biological integration in great detail, such as the
genome or proteome, but it is in the higher-order spatial and temporal patterns
of cellular (and beyond) dynamics where more answers to our questions about
human pathological processes can be found. Complex disease processes express themselves in spatial and temporal
patterns at higher-order levels of biological integration beyond the genome
and proteome level. Genes and proteins are a fundamental part of the process, but the
entire process is not represented in it entirety at the genome or proteome level.
Genes are a part of a disease process, but a disease process is not entirely
contained within (a) gene(s). Increasing our understanding about molecular
processes on a cytomic scale is required to get a grip on complex patho-physiological
processes. We have to build better bridges from basic research to clinical
applications.
Higher order biological phenomena make a significant contribution to the pathway
matrix from gene to disease and vice versa. However, these higher-order levels of biological integration are still
being studied is a dispersed way, due to the enormous technological and
scientific challenges we are facing. Several initiatives are underway
(e.g. the Physiome Project), but
crosslinking our research from genome to organism, from submicron to gross anatomy and physiology
through an improved understanding of cellular processes at the cytome level is still missing in my opinion.
Understanding cellular
patho-physiology requires exploration of cellular spatial
and temporal dynamics, which can be done either in-vitro or in-vivo.
Understanding biological processes and diseases requires an understanding of the structure and function
of cellular processes. Two approaches allow for large scale exploration of cellular phenomena,
High Content Screening and
Molecular Imaging.
Cellular structure and function is accessible with
multiple technologies, such as microscopy (extracorporal
and intracorporal), flow cytometry, magnetic
resonance imaging (MRI), positron emission tomography (PET), near-infrared
optical imaging, scintigraphy, and autoradiography,
etc. (Heckl S, 2004). Whatever allows us a view on
the human cytome wil help us to improve our
understanding of human diseases, both for diagnosis as well as for treatment.
We must gather more (quantitative) and better (predictive) information at
multiple biological levels of biological integration to improve our
understanding of human biology in health and disease.
The physiome
is the quantitative description of the functioning organism in normal and pathophysiological states (Bassingthwaighte
JB, 2000). A living organism and its cells are high-dimensional systems, both
structural and functional. A 4-D physical space (XYZ, time) is still a
formidable challenge to deal with compared to the 1-D problem of a
DNA-sequence. The even higher-order feature hyperspace which is derived from
this 4-D space is even further away from what we can easily comprehend. We
focus the major efforts of our applied research on the level of technology we
can achieve, not on the level of spatial and temporal understanding which is
required. Applied research is suffering from a scale and dimensionality deficit
in relation to the physical reality it should deal with. Reality does not
simplify itself to adapt to the technology we use to explore biology just to
please us.
Deliverables of a Human Cytome Project
The outcome of cytome research and a Human Cytome Project (HCP) should improve our
understanding of in-vivo patho-physiology in man.
It should:
- Reduce attrition in clinical development
- Improve predictive power of preclinical-clinical transition
- Improve predictive power of preclinical disease models
- Improve predictive power of early diagnosis of disease
- Improve predictive power of disease models
We must achieve a better understanding of disease processes in:
- Physiologicaly relevant cellular models
- The cytomes of model organisms
- The entire human cytome
What should we do to achieve this? Study pathological processes in:
- Physiologicaly relevant cellular models
- Study the cytomes of model organisms and man in-vitro and in-vivo
- Build a better correlation matrix between models and man
A Human Cytome Project (HCP) shines light on multiple levels of the trajectory from basic science to clinical applications:
- Basic Research: directed towards fundamental understanding of biology and disease processes
- Translational Research: concerned with moving basic discoveries from concept into clinical evaluation
- Critical Path Research: directed toward improving the product development process by establishing new evaluation tools
The rest of this article will discuss the key issues and the scientific foundation to achieve this. The technology and science we need is (almost) there, we must put it to good use.
Exploring the biology of a system
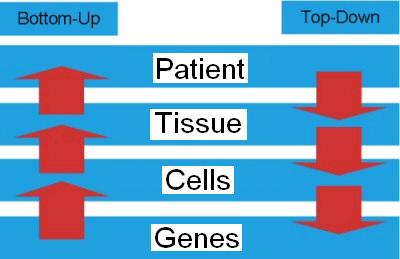
Figure 2. Exploring a biological system from top to bottom or bottom to top. |
We want to achieve a high inner resolution (detail) but at the same time we want to have a large
outer resolution (overview). We want to study the entire biological
system, but at a molecular resolution (Selinger DW, 2003).
We could call this molecular physiology.
To understand the structure and function of a (biological) system can be done in two ways or directions. Systematicaly
studying the behavior of a biological system can be done bottom-up and top-down (Figure 2). We can look at the emerging
properties of networks derived from molecules or we can look at the properties of molecules derived from
networks. Let us compare the bottom-up approach of systems biology (system of biology) with the top-down approach of the
exploration of the cytome (biology of the system):
Systems biology is an integrated way, using computational models and pathway interference
(experiments), of studying the effects of individual pathways (or drugs, targeting these pathways)
on the behaviour of a whole organism or an organ/tissue.
Studying the biology of a system is an integrated way, using computational models and pathway interference
(experiments), of studying the molecular function and phenotype of a whole organism or an organ/tissue
and its effects on individual pathways (or drugs, targeting these pathways).
Systems biology and the biology of the system both meet and merge at the level of
the dynamic network of interacting pathways.
The dynamics of cellular systems
can be explored in a global approach, which is now known as
systems biology.
Systems biology is not the biology of systems, it is the region between the
individual components and the system. It deals with those emerging properties
that arise when you go from the molecule to the system.
Systems biology is the in-between
between physiology or holism, which study the entire system, and molecular biology,
which only studies the molecules (reductionist approach). As such systems biology is
the glue between the genome and proteome on one side and the cytome and physiome
on the other side. The top-down approach of cytome and physiome research and the bottom-up
approach of genome and proteome research meet each other in systems biology.
I took me a while to come to terms
with systems biology, as I was trained (eighties of the 20th century) in medicine and
molecular biology in a traditional way. Systems biology studies
biological systems systematically and extensively and in the end tries to
formulate mathematical models that describe the structure of the system (Ideker
T., 2001; Klapa MI, 2003; Rives A.W, 2003). The end-point of present day systems biology
only takes into account infra-cellular dynamics and leaves iso- and epi-cellular
phenomena to "physiology". A "systems", but top-down, approach to
cytomics and physiomics is feasible with the technologies which are now emerging
(e.g. HCS, HCA, fluorescent probes, biomarkers, molecular imaging,..).
Studying the physics and chemistry of protein interactions cannot ignore the spatial and temporal
dynamics of cellular processes. We study nature "horizontaly", e.g. the genome
or proteome, while the flux in nature goes "verticaly", through a web of
intertwined pathways evolving in space and time. The focus of traditional -omics research
(genomics, proteomics) is perpendicular to the flow of events in nature.
The resultant vector which signifies our understanding of nature is aligned with the
way we work, not with the true flow of events in nature.
Molecular taxonomy or systems biology (genomics, proteomics) will not provide us
with all the answers we need to know, it is however an important stepstone
from molecule to man.
How to explore and find
new directions for research
Biology is inherently non-linear and complex.
Many current models in biology are simplified linear approximations of reality, often derived to
fit into available technological resources. At the moment we still expect
that an oligo- or even mono-parametric low-dimensional analysis will allow us
to draw conclusions with sufficient predictive power to start working ourselves all the way up to
the disease processes in an entire organism. We are still using quite a few disease
models with a predictive deficit, which allow us to gather data at great speed and quantity,
but in the end the translation of the results into efficient treatment of
diseases fails in the majority of cases (up to 90% attrition in clinical development).
The cost of this inefficient process is becoming a burden, which both society
and the pharmaceutical industry will not be able to support indefinitely.
As "the proof is in the pudding", not in its ingredients, we have to
improve the productivity of biomedical and pharmaceutical research and broaden
our functional understanding of disease processes in order to prepare ourselves
for the challenges facing medicine and society.
If there were no
consequences on the speed of exploration in relation to the challenges medicine
is facing today, the situation would of course be entirely different. In many
cases, the formulation of an appropriate hypothesis is very difficult and the
resulting cycle of formulating a hypothesis and verifying it is a slow and
tedious process. In order to speed up the exploration of the cytome, a more
open and less deterministic approach will be needed (Kell
DB, 2004).
Analytical tools need to be
developed which can find the needle in the haystack, without a priori knowledge
or in other words we should be able to find the black cat in a dark room,
without knowing or assuming that there is a black cat. An open and
multi-parametric exploration of the cytome should complement the more
traditional hypothesis driven scientific approach, so we can combine speed with
in-depth exploration in a two-leveled approach to cytomics. The
multi-dimensional and multi-scale "biological space" which we need to deal
with, requires a more multi-factorial exploration than the way we explore the
"biological space" at this moment. Understanding complexity cannot
be achieved without facing complexity.
Many disease models have proven
their value in drug discovery and pre-clinical development as we can withess in our daily
lives by all the drugs which cure many diseases which were once fatal or debilitating.
But we are now facing diseases which require a beter understanding of biological complexity
than ever before. We still close our eyes to much of the complexity we observe; because
our basic and applied disease models in drug discovery and development are not up to the
challenge we are facing today. We reduce the complexity of our disease models below the limits
of predictive power and meaningfulness. We must reduce the complexity of possible conclusions
(improvement or deterioration), but not the quality of process feature representation or
data extraction into our mathematical models. The value of a disease model does
not lie in the technological complexity of the machinery we use to study it,
but in its realistic representation of the disease process we want to mimic. The model is
derived from reality, but reality is not derived from the model.
A disease model in drug discovery or
drug development which fails to generate data and conclusions which hold into clinical development,
years later, fails to fulfill its mission. Disease-models are not meant to predict future
behavior of the model, but to predict the outcome of a disease and a treatment outside the model.
The residual gap between the model and the disease, in complex diseases, is in many cases too big to
allow for valid conclusions out of experiments with current (low-level) disease
models. Due to deficient early-stage disease models, the attrition rate in
pharmaceutical research is unacceptably high (80 to 90% or 4 out of 5 drugs in
clinical development). Translation of the results from drug discovery and preclinical
development into clinical success fails in about 90% of all developmental drugs
Every physical or
biological system we try to explore shows some background variation which we
cannot capture into our models. We tend to call this unaccounted variation
background noise and try to eliminate it, by randomization of experiments or
simply close our eyes for it. The less variation we are capable to capture into
our models, the more vulnerable we are for losing subtle correlations between
events. It is our inability to model complex space-time dynamics which makes us
stick to simplified models which suffer from a correlation deficit in relation
to reality. Biological reality does not simplify itself just to please us, but
we must adapt ourselves to the dynamics of biological reality in order to
increase the correlation with in vivo processes.
It is often said that the
easy targets to treat are found already, but in relation to the status of
scientific knowledge and understanding, "targets" were never easy to find.
Disease models were just inadequate to lead to an in-depth understanding of the
actual dynamics of the disease process. Just remember the concept of
"miasma"
before the work of Louis Pasteur and Robert Koch on infectious diseases. Only
when looking back with present day knowledge we declare historical research as
"easy", but we tend to forget that those scientists were fighting an uphill
battle in their days. We are now facing new challenges in medicine and drug discovery
for which we need a paradigm change in our approach to deal with basic and
applied research.
Instead of focusing on ever
further simplifying our low-dimensional and oligo-parametric disease models in
order to speed them up and only increasing the complexity of the machinery to
study them, we need a paradigm shift to tackle the challenges ahead of us.
Increasing quantity with unmatched quality of correlation to clinical reality
leads to correlation and predictive deficits. Understanding biological complexity,
means capturing its complexity in the first place. We have to create a quantitative
hyperspace derived from high-order spatial and temporal observations
(manifold) to
study the dynamics of disease processes entire cytomes and organisms. The
parameterization of the observed physical process has to represent the
high-dimensional (5D, XYZ, time, spectrum) and multi-scale reality underlying
the disease process. Each physical or feature space can be given a
coordinate system
(Cartesian, polar, gauge) which puts individual objects and processes into a
relative relation to each other for further quantitative exploration.
Homo siliconensis
Gathering more and better
quality information about cytomic processes, will hopefully allow us to
improve disease models up to a point where improved in-silico models will help
us to complement in-vivo and in-vitro disease models (Sandblad
B, 1992; Bassingthwaighte JB., 1995; Bassingthwaighte JB, 2000; Higgins G, 2001; Loew LM, 2001; Slepchenko BM,
2003; Takahashi, K., 2003; Berends M, 2004; De Schutter
E, 2004).
Cellular models are being built through the inclusion of a broad spectrum of processes
and a rigorous analysis of the multiple scale nature of cellular dynamics (Ortoleva P, 2003; Weitzke EL, 2003).
Modeling of spatially distributed biochemical networks can be used to model the
spatial and temporal organization of intracellular processes (Giavitto JL, 2003).
The physiome
is the quantitative description of the functioning organism in normal and pathophysiological states (Bassingthwaighte
JB, 2000). Gradually building the Homo (sapiens) siliconensis
or in-silico man will allow us to study and validate our disease models at
different levels of biological organization. Building a rough epi-cellular model, based on our knowledge of physiology
and gradually increasing the spatial and temporal functional resolution of the
model by increasing its cellularity could allow for
improving our knowledge and understanding on the way to a full-fledged
in-silico model of man. (Infra-) Cellular resolution is not needed in all
cases, so the model should allow for dynamic up- and down-scaling its
granularity of structural and functional resolution in both space and time.
Global, low-density models could be supplemented by a patchwork of highly
defined cellular models and gradually merge into a unified multi-scale dynamic
model of the spatial and temporal organization of the human cytome.
What to do and the way to go?
The goal of a Human Cytome Project
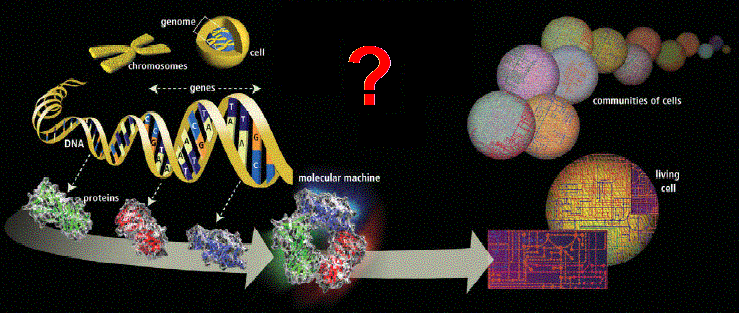
Figure 3. There is a lot of complex activity needed to build a complex cellular system (cytome) from its genes. Source: HGP media |
The functional and
structural characterization (spatial, temporal) of the processes and structures
leading to the phenotypical expression of the (human) cytome in a quantitative
way is in my opinion the ultimate goal of an endeavor on the scale of a
Human Cytome Project (HCP). We should reach a point where
we are able to understand (complex) disease processes and design disease models
which are capable to capture the multifactorial complexity of the in-vivo in-organism
dynamics of (a) disease processes with high predictive power and correlation to
clinical reality in the human biosystem (Figure 3).
This knowledge should be
made broadly available for the improvement of diagnostics, disease treatments
and drug discovery. It is the prerequisite to come to a better understanding of
disease processes and to develop and improve treatments for new, complex and
life threatening diseases for which we do not find an answer with our current
genome and proteome oriented approach only.
Studying the Cytome
First try to walk and then
run. Studying the (human) cytome as such is basically another way of looking at
research on cellular systems. We go from a higher level of biological
organization (cytome) to a lower one (proteome and cytome). Any research which
starts from the molecular single cell phenotypes in combination with exhaustive
bioinformatics knowledge extraction, is cytomics (Valet G, 2003). The only thing you need is something like a
flow-cytometer or a (digital) microscope to extract
the appropriate datasets to start with. Molecular Imaging
is also within reach for exploring disease processes in-vivo.
This approach can be used for for basic research, drug discovery,
preclinical development or even clinical development. Generating cytome-oriented
data and getting results is within reach of almost every scientist and lab.
Increasing the throughput may be required
for industrial research and for a large scale project, but this is not always
necessary for a Proof Of Concept (P.O.C.) or for studying a specific subtopic.
To explore and understand biological processes in-vitro we can use
High Content Screening (HCS) and
High Content Analysis (HCA). To study cellular processes in-vivo we can use
Molecular Imaging in model organisms and in man.
We already have the technology at hand to "bridge the gap from bench to bedside"
with translational research.
Organizational aspects
To study the entire human
cytome will require a broad multidisciplinary a multinational approach, which
will involve scientists from several countries and various disciplines to work
on problems also from a functional and phenotypical point of view and top-down,
instead of bottom-up. Both academia and industry will have to work together to
avoid wasting too much time on scattered efforts and dispersed data. The
organizational complexity of a large multi-center project will require a
dynamic management structure in which society (politicians), funding agencies,
academia and the industry participate in organizing and synchronizing the
(inter)national effort (e.g. NIH Roadmap,
EU DG Research,...). Managing and
organizing such an endeavor is a daunting task and will require excellent
managerial skills from those involved in the
process, besides their scientific expertise (Collins F.S., 2003b).
The challenges of a
Human Cytome Project will not allow us to concentrate on only a few techniques or
systematically describing individual components, but we must keep a broad
overview on cellular system genotype-phenotype correlation by multi-modal
(system-wide) exploration. Increasing our understanding about molecular
processes on a cytomic scale is required to get a grip on complex patho-physiological
processes.
We will need an open systems design in order to be able to exchange data and
analyze them with a wide variety of exploratory and analytical tools in order
to allow for creating a broad knowledgebase and proceed with the exploration of
the cytome without wasting too much time on competing paradigms and scattered data.
"The proponents of competing paradigms tend to talk at cross purposes - each paradigm
is shown to satisfy more or less the criteria that it dictates for itself and to fall
short of a few of those dictated by its opponent"
Thomas S. Kuhn, 1962).
The project should be
designed in such a way that along the road intermediate results would already
provide beneficial results to medicine and drug development. Intermediate
results could be derived from hotspots found during the process and worked out
in more detail by groups specializing in certain areas. As such the project
could consist of a large scale screening effort in combination with specific
topics of immediate interest. The functional exploration of pathways involved
in pathological processes, would allow us to proceed faster towards an
understanding of the process involved in a disease. It is best to take a dual
approach for the project, which on one side focuses on certain important diseases
(cancer, AD �), and on the other side a track which focuses on cellular
mechanisms such as cell cycle, replication, cell type differentiation (stem
cells).� The elucidation of these
cellular mechanisms, will lead to the identification of hot-spots for further
research in disease process and allow for the development of new therapeutic
approaches.
A strategy for observation
We manipulate and observe a process in its native
environment, i.e. in its complex biological context. Therefore we must be able to observe a
high dimensional environment (3D spatial plus time and spectrum) with a high inner
(spatial and temporal resolving power) and wide outer resolution (field of view).
No single technology will deliver all in all circumstances. A device can sample a
physical space (XYZ) at a certain inner and outer resolution, which translates
in a cellular system into a structure space, such as the nucleus, Golgi,
mitochondria, lysozomes, membranes, organs, etc. We can sample a time axis also
within a certain inner and outer resolution, which in a cellular system
translates in life cycle stages such as cell division, apoptosis and cell
death. The spectral axis (electromagnetic spectrum) is used to discriminate
between spatially and temporally coinciding objects. It is used by means of
artificially attached labels which allow us to use spectral differentiation to
identify cellular structures and their dynamics. It expands the differentiating
power of the probing system. Expanding our observations to entire organisms
while retaining high resolving power is the next step (e.g. moleculer imaging).
We use a combination of
space, time and spectrum to capture and differentiate structures and processes
in and around the cells of the human cytome. The cytome is built from all
different cells and cell-types of a multi-cellular organism so we multiplex our
exploration over multiple cells and cell types, such as hepatocytes,
fibroblasts, etc.
In the cells of the human
cytome we insert structural and functional watchdogs (reporters) on different
life-cycle time points and into different organelles and around cells (Deuschle K, 2005). At the moment we already have a
multitude of reporters available to monitor structural and functional changes
in cells (fluorescent probes, biomarkers, ...).
This inserts a sampling grid or web into cells which will report structural and functional changes which we can use as
signposts for further exploration. We turn cells into 4D arrays or grids for
multiplexing our observations of the spatial and temporal changes of cellular
metabolism and pathways. It is like using a 4D �spiderweb�
to capture cellular events. Instead of extracting the 4D matrix of cellular
structure and dynamics into 2D microarrays (DNA,
protein �), we insert probe complexes into the in-vivo intracellular space-time
web. We create an intracellular and in-vivo micro-matrix or micro-grid.
Structural and functional
changes in cells will cause a space-time "ripple" in the structural
and functional steady state of the cell and if one of the reporters is in
proximity of the status change it will provide us with a starting point for
further exploration. A living cell is not a static structure, buth an oscillating complex of both structural and
functional events. The watchdogs are the bait to capture changes and act as
signposts from which to spread out our cytome exploration. We could see them as
the starting point (seeds) of a shotgun approach or the threads of a spiderweb for cytome exploration.
The spatial and temporal
density and sensitivity of our reporters and their structural and functional
distribution throughout the cytome will define our ability to capture small
changes in the web of metabolic processes in cells. At least we capture changes
in living cells (in-vivo or in-vitro), closely aligned with the space-time
dynamics of the physilogicla process. We should try to align the watchdogs with hot-spots of cellular structure and
function. The density and distribution of watchdogs is a dynamic system, which
can be in-homogeneously expanded or collapsed depending on the focus of
research.
Technology for cell and cyctome manipulation
Multiple techniques are available to maniplulate the dynamics of cellular systems,
either for single cells or for the entire cytome of an organism, e.g. RNAi, chemical genomics,
morpholinos, aptamers, etc. . By monitoring the functional and structural changes we can learn
about the underlying in vivo processes in a complex biosystem.
RNA interference (RNAi) can
silence gene expression and can be used to inhibit the function of any chosen
target gene (Banan M, 2004; Campbell TN, 2004; Fire
A., 1998; Fraser A. 2004; Mello CC, 2004; Mocellin,
2004). Large scale RNAi creening is now within reach
(Sachse C, 2005). This technique can be used to study
the effect of in-vivo gene silencing on the expressed phenotype (watchdog
monitoring) in a transient way, both in individual cells as well as in
organisms.
Aptamers
are small molecules that can bind to another molecule (Ellington, 1990). We can use
DNA or RNA aptamers and protein aptamers to interfere with cellular processses (Crawford M, 2003; Toulme, 2004).
Morpholino oligos are used to block access of other molecules to
specific sequences within nucleic acid molecules (Ekker SC., 2000. They can block access of other molecules to small
(~25 base) regions of ribonucleic acid (RNA). Morpholinos are sometimes referred to as PMO,
an acronym for phosphorodiamidate morpholino oligo (Summerton J., 1999; Achenbach TV, 2003).
Chemical genetics is the study of biological systems using small molecule ('chemical')
intervention, instead of only genetic intervention. Cell-permeable and selective small
molecules can be used to perturb protein function rapidly, reversibly and conditionally
with temporal and quantitative control in any biological system (Spring DR., 2005; Haggarty SJ. 2005).
Stem cells can be made to
differentiate into different cell types and the differentiation process montired for spatial and temporal changes and
irregularities. By using stem cells we can mimic (and avoid) taking multiple
biopsies at different life stages of an individual and its cells. The resulting
cell types can be used for multiplexing functional and structural research of
intracellular processes. RNAi can be applied to stem cell research to study
stem cell function (Zou GM, 2005).
Technology for observation
We need technology (probes and detectors) to monitor and manipulate a complex biological system, ranging from
mono-cellular up to complete cytomes (organisms).
Human biology can be
explored at multiple levels and scales of biological organization, by using
many different techniques, such as CT, MRI, PET, LM, EM, etc. each providing us with
a structural and functional subset of the physical phenomena going on inside
the human body. In this section I will focus on the cellular level and
on microscopy-based techinques and Molecular Imaging techniques.
Achieving (sub-) cellular resolution poses thechnological
challenges, which can be met by the combined use of multiple techniques and instruments.
Non-invasive molecular imaging modalities such as optical imaging (OI), magnetic resonance imaging (MRI), MR spectroscopy and positron emission tomography (PET) allow in-vivo assessment of metabolic changes in animals and humans. While CT and MRI provide anatomical information, optical fluorescence and bioluminescence imaging and especially PET reveal functional information, in case of PET even in the picomolar range.
The necessity to explore
the cellular level of the human physiome poses some
demands on the spatial, spectral and temporal inner and outer resolution which
has to be met by the technology used to extract content from the cell. However
there is no one-on-one overlap between the biological structure and activity at
the level of the cytome and our technological means to explore this level. Life
does not remodel its physical properties to adapt to our exploratory
capabilities. The alignment of the scale and dimensions of cellular physics
with our technological means to explore is still far from perfect. The
discontinuities and imperfections in our exploratory capacity are a cause of
the fragmentation of our knowledge and understanding of the structure and
dynamics of the cytome and its cells. Our knowledge is aligned with our
technology, not with the underlying biology.
Cellular system observation - image based cytometry
Every scientific challenge
leads to the improvement of existing technologies and the development of new technologies
(Tsien R, 2003). Technology to explore the cytome is already available today
and exciting developments in image and flow based cytometry are going on at the
moment. The dynamics of living cells is now being studied in great detail by
using fluorescent imaging microscopy techniques and many sophisticated light
microscopy techniques are now available (Giuliano KA,
1998; Tsien RY, 1998; Rustom A, 2000; Emptage NJ., 2001; Haraguchi T.
2002; Gerlich D, 2003b; Iborra
F, 2003; Michalet, X., 2002; Michalet,
X., 2003; Stephens DJ, 2003; Zimmermann T, 2003). Studying intra-vital
processes is possible by using microscopy (Lawler C, 2003). Quantitative microscopy requires a
clear understanding of the basic principles of digital microscopy and sampling
to start with, which goes beyond the principles of the Nyquist sampling theorem
(Young IT., 1988).
Advanced microscopy
techniques are available to study the morphological and temporal events in
cells, such as confocal and laser scanning microscopy (LSM), digital
microscopy, spectral imaging, Fluorescence Lifetime Imaging Microscopy (FLIM),
Fluorescence Resonance Energy Transfer (FRET) and Fluorescence Recovery After Photobleaching (FRAP) (Cole, N. B. 1996; Truong K, 2001, Larijani B, 2003; Vermeer JE, 2004). Spectral imaging microscopy and
FRET analysis are applied to cytomics (Haraguchi T,
2002; Ecker RC, 2004). Fluorescent speckle microscopy
(FSM) is used to study the cytoskeleton in living cells (Waterman-Storer CM, 2002; Adams MC, 2003; Danuser
G, 2003).
Laser scanning (LSM) and
wide-field microscopes (WFM) allow for studying molecular localisation and
dynamics in cells and tissues (Andrews PD, 2002). Confocal and multiphoton
microscopy allow for the exploration of cells in 3D (Peti-Peterdi
J, 2003). Multiphoton microscopy allows for studying the dynamics of spatial,
spectral and temporal phenomena in live cells with reduced photo toxicity
(Williams RM, 1994; Piston DW, 1999; Piston DW. 1999b; White JG, 2001).
Green fluorescent protein
(GFP) expression is being used to monitor gene expression and protein
localization in living organisms (Shimomura O, 1962; Chalfie
M, 1994; Stearns T. 1995; Lippincott-Schwartz J,
2001; Dundr M, 2002; Paris S, 2004). Using GFP in
combination with time-resolved microscopy allows studying the dynamic
interactions of sub-cellular structures in living cells (Goud
B., 1992; Rustom A, 2000). Labelling of bio-molecules
by quantum dots now allows for a new approach to multicolour optical coding for
biological assays and studying the intracellular dynamics of metabolic
processes (Chan WC, 1998; Han M, 2001; Michalet, X.,
2001; Chan WC, 2002; Watson A, 2003; Alivisatos, AP,
2004; Zorov DB, 2004).
The resolving power of
optical microscopy beyond the diffraction barrier is a new and interesting
development, which will lead into so-called super-resolving fluorescence
microscopy (Iketaki Y, 2003). New microscopy
techniques such as standing wave microscopy, 4Pi confocal microscopy, I5M
and structured illumination are breaking the diffraction barrier and allow for
improving the resolving power of optical microscopy (Gustafsson
MG., 1999; Egner, A., 2004). We are now heading
towards fluorescence nanoscopy, which will improve spatial resolution far below
150 nm in the focal plane and 500 nm along the optical axis (Hell SW., 2003;
Hell SW, 2004).
Exploring ion flux in
cells, such as for Calcium, is already available for a long time (Tsien R,
1981, Tsien R 1990; Cornelissen, F, 1993). Locating the spatial and temporal
distribution of Ca2+ signals within the cytosol
and organelles is possible by using GFP (Miyawaki A,
1997). Fluorescence ratio imaging is being used to study the dynamics of
intracellular Ca2+ and pH (Bright GR, 1989; Silver RB., 1998; Fan
GY, 1999; Silver RB., 2003; Bers DM., 2003).
Microscopy is being used to
study Mitochondrial Membrane Potentials (MMP) and the spatial and temporal
dynamics of mitochondria (Zhang H, 2001; Pham NA, 2004). The distribution of H+
ions across membrane-bound organelles can be studied by using pH-sensitive GFP
(Llopis J, 1998)
Electron Microscopy allows
studying cells almost down to the atomic level. Atomic Force Microscopy (AFM)
allows studying the structure of molecules (Alexander, S., 1989; Drake B, 1989;
Hoh, J.H., 1992; McNally HA, 2004). Multiple techniques
can be used, such as combining AFM for imaging living cells and compare this
with Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy
(TEM) (Braet F, 2001).
High Content Screening
(HCS) is available for high speed and large volume screening of protein
function in intact cells and tissues (Van Osta P., 2000; Van Osta P., 2000b; Liebel U, 2003; Conrad C, 2004; Abraham VC, 2004; Van Osta
P., 2004). New research methods are bridging the gap between neuroradiology and neurohistology,
such as magnetic resonance imaging (MRI), positron emission tomography (PET),
near-infrared optical imaging, scintigraphy, and
autoradiography (Heckl S, 2004).
Processes can be monitored by using bioluminescence to monitor gene expression in living mammals
(Contag CH, 1997; Zhang W., 2001). Both bioluminescence and fluorescence technology can be used
for studying disease processes and biology in vivo (Choy G, 2003). Multimodality reporter
gene imaging of different molecular-genetic processes using fluorescence,
bioluminescence (BLI), and nuclear imaging techniques is also feasible (Ponomarev V, 2004).
Cellular system observation - flow based cytometry
Flow Cytometry allows us to
study the dynamics of cellular processes in great detail (Perfetto SP, 2004; Voskova
D, 2003; Roederer M, 2004). Interesting developments are leading to fast
imaging in flow (George TC, 2004). Combining both image and flow based
cytometry can shed new light on cellular processes (Bassoe
C.F., 2003).
Cytome wide system observation - molecular imaging
In recent years tools came available for establishing the actions of agents in the complex biochemical networks characteristic of fully assembled living systems (Fowler BA., 2005; Turner SM, 2005). Molecular Imaging is broadly defined as the characterization and measurement of biological processes in living animals at the cellular and molecular level. Noninvasive "multimodal" in vivo imaging is not just becoming standard practice in the clinic, but is rapidly changing the evolving field of experimental imaging of genetic expression ("molecular imaging"). The development of multimodality methodology based on nuclear medicine (NM), positron emission tomography (PET) imaging, magnetic resonance imaging (MRI), and optical imaging is the single biggest focus in many imaging and cancer centers worldwide and is bringing together researchers and engineers from the far-ranging fields of molecular pharmacology to nanotechnology engineering. The rapid growth of in vivo multimodality imaging arises from the convergence of established fields of in vivo imaging technologies with molecular and cell biology.
The role of any multimodal imaging approach ideally should provide the exact localization,
extent, and metabolic activity of the target tissue, yield the tissue flow and function or functional changes
within the surrounding tissues, and in the topic of imaging or screening, highlight any pathognomonic changes
leading to eventual disease.
High-resolution scanners are bing used for imaging small animals of use in the many new molecular
imaging centers appearing worldwide for reporter gene-expression imaging studies in small animals.
The coupling of nuclear and optical reporter genes represents only the beginning of far wider applications
of this research. Initially, fluorescence and bioluminescence optical imaging, in providing a cheaper
alternative to the more intricate microPET, microSPECT, and microMRI scanners, have gathered the most
focus to date. In the end, however, the volumetric tomographic technologies, which offer deep tissue
penetration and high spatial resolution, will be married with noninvasive optical imaging.
Molecular Imaging (will) bridge(s) the gap from (sub-)cellular to macrocellular anatomy and physiology,
allowing us to study the dynamics of cellular process in-vivo (Kelloff GJ, 2005; Kola I, 2005).
The ability to accurately measure the in-vivo molecular response to a drug allows to demonstrate the pharmacological effects in a smaller number of individuals and at lower doses. This forms the basis for the Proof-of-Concept (POC) trials that aim to confirm and describe the desired pharmacodynamic (PD) effect in man before starting traditional clinical trials. Changes in molecular markers can often be demonstrated at a fraction of the therapeutic dose. Microdosing is a technique whereby experimental compounds are administered to humans in very small doses (typically 1/100th of a therapeutic dose) (Wilding IR, 2005). The drug's pharmacokinetics are characterized through the use of very sensitive detection techniques, such as accelerator mass spectrometry (AMS) and PET (Bergstrom M, 2003). Human microdosing aims to reduce attrition at Phase I clinical trials.
Cell and cytome analysis - image analysis
In order to come to come to
a quantitative understanding of the dynamics of in-vivo cellular processes
image processing, methods for object detection, motion estimation and
quantisation are required. The first step in this process is the extraction of
image components related to biological meaningful entities, e.g. nuclei,
organelles etc., Secondly quantitative features are applied to the selected
objects, such as area, volume, intensity, texture parameters, etc. Finally a
classification is done, based on separating, clustering, etc. of these
quantitative features.
New image analysis and
quantification techniques are constantly developed and will enable us to
analyze the images generated by the imaging systems (Van Osta P, 2002; Eils R, 2003; Nattkemper TW,
2004; Wurflinger T, 2004). The quantification of
high-dimensional datasets is a prerequisite to improve our understanding of
cellular dynamics (Gerlich D, 2003; Roux P, 2004; Gerster AO, 2004). Managing tools for classifiers and
feature selection methods for elimination of non-informative features are being
developed to manage the information content and size of large datasets (Leray, P., 1999; Jain, A.K., 2000; Murphy, R.F., 2002;
Chen, X., 2003; Huang, K., 2003; Valet GK, 2004)
Imaging principles based on
physics and human vision principles allow for the development of new and
interesting algorithms (Geusebroek J.M., 2001; Geusebroek J. M., 2003; Geusebroek
J. M., 2003b; Geusebroek J. M., 2005). The necessary
increase of computing power requires both a solution at the level of
computation as increasing the processing capacity (Seinstra
F.J., 2002;
The development of new and
improved algorithms will allow us to extract quantitative data to create the
high-dimensional feature spaces for further analysis.
Exploring cells and cytomes on a large scale
This section will be
expanded in a separate document on a Research Management System (RMS).
My personal interest was to
build a framework in which acquisition, detection and quantification are
designed as modules each using plug-ins to do the actual work (Van Osta P,
2004) and which in the end could manage a truly massive exploration of the human
cytome. Two approaches allow for large scale exploration of cellular phenomena,
High Content Screening and
the emerging field of Molecular Imaging
The basic principle of a
digital imaging system is to create a digital in-silico representation of the
spatial, temporal and spectral physical process which is being studied. In
order to achieve this we try to let down a sampling grid on the biological
specimen. The physical layout of this sampling grid in reality is never a
precise isomorphic cubical sampling pattern. The temporal and spectral sampling
inner and outer resolution is determined by the physical characteristics of the
sample and the interaction with the detection technology being used.
The extracted objects are
sent to a quantification module which attaches an array of quantitative
descriptors (shape, density �) to each object. Objects belonging to the same
biological entity are tagged to allow for a linked exploration of the feature
space created for each individual object (Van Osta P., 2000; Van Osta P.,
2002b, Van Osta P., 2004). The resulting data arrays can be fed into analytical
tools appropriate for analysing a high dimensional linked feature space or
feature hyperspace.
Data analysis and data management
Managing and analyzing
large datasets in a multidimensional linked feature space or hyperspace will
require a change in the way we look at data analysis and data handling.
Analyzing a multidimensional feature space is computationally very demanding
compared to a qualitative exploration of a 3D image.
We often try to reduce the complexity of our datasets before we feed them into
analytical engines, but sometimes this is a �reductio
ad absurdum�, below the level of meaningfulness. We have to create tools to be
able to understand high-dimensional feature �manifolds� if we want to capture
the wealth of data cell based research can provide. Transforming a
high-dimensional physical space into an even higher order feature space
requires an advanced approach to data analysis.
The conclusion of an
experiment may be summarized in two words, either �disease� or �healthy�, but
the underlying high-dimensional feature space requires a high-dimensional
multiparametric analysis. Data reduction should only occur at the level of the
conclusion, not at the level of the quantitative representation of the process.
The alignment of a feature manifold with the multi-scale and multidimensional
biological process would allow us to capture enough information to increase the
correlation of our analysis with the space-time continuum of a biological
process.
Building the
multidimensional matrix of the web of cross-relations between the different
levels of biological organization, from the genome, over the proteome, cytome
all the way up to the organism and its environment, while studying each level
in a structural (phenotype) and functional way, will allow us to understand the
mechanisms of pathological processes and find new treatments and better
diagnostics tools. A systematic descriptive approach without a functional
complement is like running around blind and it takes too long to find out about
the overall mechanisms of a pathological process or to find distant
consequences of a minute change in the pathway matrix.
We should also get serious
on a better integration of functional knowledge gathered at several biological
levels, as the scattered data are a problem in coming to a better understanding
of biological processes. The current data storage models are not capable of
dealing with heterogeneous data in a way which allows for in-depth
cross-exploration. Data management systems (data warehouses)
will need to broaden their scope in
order to deal with a wide variety of data sources and models. Storage is not
the main issues, the use and exploration of heterogeneous data is the
centerpiece of scientific data management. Data originating from different
organizational levels, such as genomic (DNA sequences), proteomic (protein
structure) and cytomic (cell) data should be linked.
Data originating from different modes of exploration, such as LM, EM, NMR and
CT should be made cross-accessible. Problems to link knowledge originating from
different levels of biological integration is mainly due to a failure of multi
scale or multilevel integration of scientific knowledge, from individual gene
to the entire organism, with appropriate attention to functional processes at
each biological level of integration.
Standardization and quality
Both basic and applied
research should adhere to the highest quality standards without sacrificing
exploratory freedom and scientific inspiration. On the experimental side,
standardization of experimental procedures and quality control is of great
importance to be able to compare and link the results from multiple
research-centers. But quality is not only a matter of experimental procedures
and instrumentation, but also of disease model validation and verifying the
congruence of a model with clinical reality.
We need to design
procedures for instrument set-up and calibration (Lerner JM, 2004). We need to
define experimental protocols (reagents�) in order to be able to compare experiments.
In addition we need to standardize data exchange procedures and standards such
as the CellML language,
CytometryML, Digital Imaging and Communications in Medicine (DICOM),
Open Microscopy Environment (OME XML)
and the Flow Cytometry Standard (FCS) (Murphy RF, 1984; Seamer
LC, 1997; Leif RC, 2003; Swedlow JR, 2003; Horn RJ.
2004; Samei E, 2004). The purpose of CellML is to
store and exchange computer-based mathematical models (Autumn A., 2003. A file format such as
the Image Cytometry Standard (ICS v.1 and 2) provides for a very flexible way to store
and retrieve multi-dimensional image data (Dean P., 1990).
Laboratory Information Manangement Systems (LIMS) and automated lab data management provide support for sample an data handling, which is an important aspect of every large scale automated research project. Laboratory Information Management Systems (LIMS) are information management systems designed specifically for the analytical laboratory. This includes research and development (R&D) labs, in-process testing labs, quality assurance (QA) labs, and more. Typically, LIMS connect the analytical instruments in the lab to one or more workstations or personal computers (PC). The rise of Informatics, coupled with the increasing speed and complexity of the analytical instruments, is driving more sophisticated data manipulation and data warehousing tools that work hand-in-glove with LIMS to manage and report laboratory data with ever greater accuracy and efficiency.
From clinical practice we could borrow some
principles of quality assurance (QA) and quality control (QC). If we want to speed up the
transfer of knowledge from basic research to clinical applications for the
benefit of mankind, we should create Standard Operating Procedures (SOPs).
Good Laboratory Practices (GLP) (FDA GLP,
OECD GLP)
would improve the quality of our methods used in the laboratory. For the instrumentation used in the research projects which
constitute a Human Cytome Project (HCP), attention should be given to the
FDA Title 21 Code of Federal Regulations
(FDA Title 21 CFR Part 11) on
Electronic Records and Electronic Signatures.
In the design and development of instrumentation, Current Good Manufacturing Practices
(cGMP) are valuable guidelines for manufacturing quality.
The Good Automated Manufacturing Practice (GAMP)
Guide for Validation of Automated Systems in Pharmaceutical Manufacture could be another source of information in
order to guarantee the quality of the data coming out of the Human Cytome Project (HCP).
The methods used for data
analysis, data presentation and visualization need to be standardized also. We
need to define quality assurance (QA) and quality control (QC) procedures and standards which can be used
by laboratories to test their procedures. A project on this scale requires a
registration and repository of cell types and cell lines
(e.g. ATCC,
ECCC). This way of working is already
implemented for clinical diagnosis, by organizations such as the
European Working Group on Clinical Cell Analysis
(EWGCCA), which could help to implement standards and procedures for a Human Cytome Project (HCP).
Reference standards for our measurements can be developed and provided by institutions such as the
Institute for Reference Materials and Measurements
(IRMM) in Europe, one of the seven institutes of the Joint Research Centre (JRC),
a Directorate-General of the European Commission (EC).
Conclusions
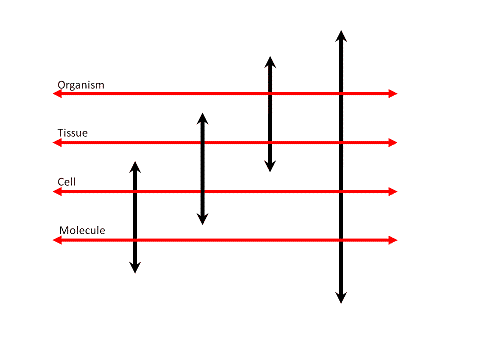
Figure 4: Exploring the organizational levels of biology.
A linked and overlapping cascade of exploratory systems
each exploring -omes at different organizational levels of biological systems
in the end allows for creating an interconnected knowledge architecture of entire cytomes.
The approach leads to the creation of an Organism Architecture (OA)
in order to capture the multi-level dynamics of an organism.
The complexity of diseases requires an improvement of our understanding of high-order biological processes in man. We cannnot close our eyes to the unmet needs of many patients, which are in need of new and better therapies. New ways to deal with disease processes are required if we want to succeed in the war against diseases threatening mankind.
References
References can be found here
Cytome Research
- Max-Planck-Institut für Biochemie
- Purdue University
- Bindley Bioscience Center at Discovery Park
- Carnegie Mellon University
- Coriell Institute
- Herzzentrum Leipzig GmbH
Additional Information
- A Human Cytome Project, posted on Monday 01 December 2003 at 10:57:46 in the bionet.cellbiol newsgroup
- Towards a Human Cytome Project
- Draft: Human Cytome Project
- Biomedical Imaging
- Image Analysis gives a clear view in research
- Application of linear scale space and the spatial color model in light microscopy
- Automated Tiled Multi-mode Image Acquisition and Processing Applied to Pharmaceutical Research
- The M5 framework for exploring the cytome
- International Human Cell Atlas Initiative
- Cell atlas project
- Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products
- New Safe Medicines Faster Project
- Priority Medicines for Europe and the World Project "A Public Health Approach to Innovation"
- Moving Medical Innovations Forward? New Initiatives from HHS
- NIH Roadmap
- EU - Research
- Cytomics
- E-Cell Project
- Physiome Project
- EuroPhysiome - STEP
- IUPS Physiome Project
- GIOME Project
- Functional genomics
- Cyttron
- National Resource for Cell Analysis and Modeling - NRCAM
- Prediction in Cell-based Systems (Predictive Cytomics)
- NCBI
- The Academy of Molecular Imaging
- The Society for Molecular Imaging
- The Crump Institute for Molecular Imaging
- Molecular Imaging Program at Stanford (MIPS)
- VCUHS Molecular Imaging Center
- Molecular Imaging Research and Clinic - K.U.Leuven
- The Data Warehousing Information Center
References
- Van Osta P, Donck KV, Bols L, Geysen J., Cytomics and drug discovery, Cytometry A. 2006 Feb 22;69A(3), pp. 117-118
- Geusebroek, J.-M., Cornelissen, F., Smeulders, A.W. and Geerts, H. (2000), Robust autofocusing in microscopy. Cytometry, 39: 1-9
- I.T. Young and L.J. van Vliet, Recursive implementation of the Gaussian filter, Signal Processing, vol. 44, no. 2, 1995, 139-151
Acknowledgments
I am indebted, for their pioneering work on automated digital microscopy and High Content Screening (HCS) (1988-2001), to my former colleagues at Janssen Pharmaceutica (1997-2001 CE), such as Frans Cornelissen, Hugo Geerts, Jan-Mark Geusebroek and Roger Nuyens, Rony Nuydens, Luk Ver Donck, Johan Geysen and their colleagues.
Many thanks also to the pioneers of Nanovid microscopy at Janssen Pharmaceutica, Marc De Brabander, Jan De Mey, Hugo Geerts, Marc Moeremans, Rony Nuydens and their colleagues. I also want to thank all those scientists who have helped me with general information and articles.
Copyright notice and disclaimer
My web
pages represent my interests, my opinions and my ideas, not those of my
employer or anyone else. I have created these web pages without any commercial
goal, but solely out of personal and scientific interest. You may download,
display, print and copy, any material at this website, in unaltered form only,
for your personal use or for non-commercial use within your organization.
Should my web pages or portions of my web pages be used on any Internet or
World Wide Web page or informational presentation, that a link back to my website (and
where appropriate back to the source document) be established. I expect at
least a short notice by email when you copy my web pages, or part of it for
your own use.
Any information here is provided in good faith but no warranty can be made for
its accuracy. As this is a work in progress, it is still incomplete and even
inaccurate. Although care has been taken in preparing the information contained
in my web pages, I do not and cannot guarantee the accuracy thereof. Anyone
using the information does so at their own risk and
shall be deemed to indemnify me from any and all injury or damage arising from
such use.
To the best of my knowledge, all graphics, text and other presentations not
created by me on my web pages are in the public domain and freely available
from various sources on the Internet or elsewhere and/or kindly provided by the
owner.
If you notice something incorrect or have any questions, send me an email.
Email: pvosta at gmail dot com
The author of this webpage
is Peter Van Osta, MD.
A first draft was published
on
Latest revision on 15 August 2008