Cytomics in Drug Discovery and Development
![[construction icon]](https://www.vanosta.be/images/worker.gif)
Index
- Introduction
- Problems of drug discovery and development - problems at several levels
- Improving drug discovery and development - attempts at several levels
- Problems with disease models - models and the intrahuman ecosystem
- Less questions, but better answers - decomplexification of questions, improving answers
- A cytome project
- A proposal for the exploration of the human cytome - only high level concept
- A framework for the exploration of the human cytome
Introduction
This webpage is meant as a humble contribution to the discussion about improving drug discovery and development, but it is still under construction. It did not go through the traditional peer review process of scientific publications and neither does it pretend to be a critical appraisal of medical evidence, so read it with care and with a critical mind. Feel free to comment and to criticise.
Why would drug discovery and development benefit from cytomics and what are the fundamental problems we face in drug discovery and development ? Cytomics and a coordinated research effort organised in a human cytome project aims at creating a better understanding of the cellular diversity of biological systems and reducing the great divide between present day reductionist models and clinical reality. Cytomics allows us to close the great divide between molecular research and the complexity of the intrahuman ecosystem. There is no clinical evidence to be found in a testtube, so the odds are against basic and applied research in our (pharmaceutical) laboratories and (pre-)clinical pipelines. Understanding the (heterogeneous) cellular level of biological organisation and complexity is (almost) within reach of present day science, which makes cytomics as a science and a human cytome project ambitious but achievable. A human cytome project is all about creating a solid translational science, not from bench to bedside, but from molecule to man. Even more than translational science, it is about transformational science as it transforms the molecular microcosm into the macrocosm of the intrahuman ecosystem and its physiology.
Although the present business model of pharmaceutical discovery and development is capable of coping with massive and late stage failures during the development life cycle of a new drug, it is not the way we should continue to work as the challenges ahead are even bigger than the ones we already faced in the past. The current business model is capable to deliver enough drugs to sustain itself, given the disease mechanisms drug discovery and development deal with, do not surpass a modest level of biological complexity. The present business model simply takes into account the high risk and failure rate of present day science. The scientific engine, from basic to applied research, of the overall process is not yet capable to predictably deliver new drugs which can stand the challenges of biological complexity. At any given moment in time the actual performance of the scientific engine of the process and its scientists has to be taken more or less for granted, and only cost (labor, headcount, process engineering, economy of scale, mergers and acquisitions) and income (price, health insurance, government pricing policies) are available for marginal improvements in process performance (efficiency) and productivity (effectiveness). Business engineering is somewhat easier than engineering the framework of science. We do what we can and what is possible, but not necessarily what is required. We look for solutions where we can find them, but not necessarily where we have to go.
We have to destroy too much capital and human effort to sustain the pipeline, which nowadays resembles a small tube in most companies (to be honest the past was not better either). We compensate for massive scientific failure rates by business model egineering. We have to build a business model on almost catastrofic failure rates compensated by high cost per product, which is only possible in medicine at this rate. But no matter how efficient we engineer our business processes, we cannot hide from the fact that we are running a pipeline process which has too much risk hidden underneath to feel comfortable with as the basis for delivering new treatments for the diseases of our society. Although the pharmaceutical process matrix is in itself consistently organised, it is is slightly out of touch with the complexity of clinical reality. This becomes visible at the end of the pipeline, when truth is finaly forced upon us and we can no longer hide from clinical reality, which extends beyond the frontiers of science. At the intermediate stages the confrontation with reality is only partial and resembles a democracy where the (clinical) opposition is (to be) excluded from voting. A safety test may prove that a candidate drug is safe, but leaves one blind on its efficacy. We cannot deal with all the biological complexity of the intrahuman ecosystem and so we have to withdraw into simplified representations of reality which are too simple as we have to acknowledge at the end of the pipeline in the vast majority of drugs being developed. Expansion of the frontiers of our knowledge is an interesting activity but the process of knowledge expansion in itself is an ineffective and inefficient process. At the intermediary steps we are capable to declare victory because we can set the rules less stringent than those which nature itself pushes through in a real world clinical situation at the end of the game. Life in the lab is therefore easier than life in the clinic, where you have to look the patient in the eye and fight diseases in reality and not in a test tube. Changing the overall process however is a Gargantuan endeavour as it is all about the core scientific engine in the first place, which is almost inert when it comes to paradigm changes.
We are doing our science right, but we are not doing the right science. The success ratio of science, when starting from basic research down to its successful application in the clinic is less than 1 percent. But as scientists have no clue which part of the 100 percent effort will succeed, society has no option but to waste 99 percent of the effort in order to succeed in 1 percent of all research (from basic research to successful application). The main problem is that our basic science is basic in dealing with biological complexity. We look at the intrahuman ecosystem through a keyhole and remain blind for most of its complexity. Although we increasingly automate the scientific process, the process in itself remains largely the same and the effectiveness does not change in the same order of magnitude as its automation which mainly drives the efficiency. We may proceed faster through some stages of the pipeline with our machines, but we still do not reach the goal in most cases. The amount of data being produced increases dramatically, but a pile of numbers does not equal a pile of true understanding of the perceived image of reality which our machines unfold to us providing the view through which we observe and perceive the disease process but still not the reality itself we would need to know and understand. Capacity does not equal performance in terms of results, activities based on the wrong assumptions do not help us in the end. Failing intermediary discovery and development stages leave us with massive failure at the final gate. We postpone the true confrontation with the complexity of nature until the end of the entire process and thereby fail to remedy the intermediary steps (pipeline science is what it is, just live with it). The moment of truth only becomes apparent during the final stages of drug development, buth then it is is already too late. We are good at scientific engineering within the boundaries of our models of biological reality, but rather inefficient in expanding the boundaries of true understanding of the dynamics of biological processes (i.e. the dynamics of biology or multilevel physiology, not just its molecular structure). Present day science, as its predecessors, is far from perfect or up to its clinical challenge, but it is all we have, so for any moment in time we just have to live with it and build our business on top of it.
The dramatic failure rate of science in the face of clinical reality is just one of the parameters in the equation of a business model. Business succeeds even when science fails massively. Pharmaceutical companies just take into account the massive failure rate of (basic) science in their business model. No matter the productivity of science, running a very profitable pharmaceutical business was, is and will always always be feasible, by compensating for the deficits of science by careful management and business engineering (which is somewhat easier than changing the paradigm of science). From the early days of industrial drug discovery and development, management techniques have been conceived and used to create a profitable business, regardless of the shortcomings of science to produce drugs which succeed in the face of clinical reality. Assembling a profitable pipeline can be done either within a traditional monolithic structure (big pharma) or a cluster of risk sharing companies each a part of the overall pipeline. These two business models establish a different risk exposure, but the overall scientific process (the actual engine which drives the pipeline) remains the same. Pharmaceutical management manages risks, which are near catastrophic in drug discovery and development. The science of conceiving the framework and managing a pharmaceutical company is even of a higher order than the scientific engine itself, as it is capable of generating profitability and bring new drugs to the people on top of a scientific process which has less chances to succeed than throwing a dice.
The question is of course, which direction we are capable to take in order to move forward; even more business engineering and automation (increase efficiency) or changing the paradigm and practice of basic and applied science (improve effectiveness), or both ? Although we are becoming increasingly efficient, we do not achieve more effectiveness in the same order of magnitude. Is academia and the industry capable to innovate its core activities ? Is changing such a complex process possible anyhow, while at the same time keeping the business in the air ? What is the value of getting more answers on the wrong questions ? What is the value of our work if the review by nature itself declines the results of our scientific effort ? Is there any hope of an improvement in the productivity of the scientific process itself ?
Do we just have to live with the as is situation of basic and applied science and make the best of it at any given moment in time ? Are we capable to improve the scientific process of knowledge expansion itself or do we continue crawling slowly at the borders of present day scientific boudaries instead of engineering the scientific process itself towards more productivity (truly succeed more often when facing the challenge of nature itself). Scientific practice is always done within the boundaries of the paradigms of the day but not along the lines demanded by nature itself. No matter how far we are today, there is something wrong with the productivity of the scientific method iself in the face of nature. The weakness is not the practice, but the process itself which is flawed towards its performance to find the right answers on complex questions. The process delivers what it is designed for by our science, but not what is required to deal with nature itself in the real clinical world in more than 90 percent of what we (try to) deliver.
As such this article is dedicated to all the patients hoping and waiting for new treatments of unmet medical needs and the improvement of existing therapies. It is also dedicated to all the scientists working in basic and applied research, working day and night to deliver these new drugs and treatments. It deals with process (technology, biology) and model deficits in drug discovery and development at various points throughout the pipeline. Pipeline analysis and engineering is a delicate process as it is required to change the bricks of the house, without destroying it.
The breakthroughs in basic research have not resulted in the creation of many new therapies for patients, which lead to the 'pipeline problem'.
Improving drug discovery and development is not a simple endeavour, as we have seen in recent years. Although this
article is critical about the (evolution of the) overall drug discovery and development process it also honours
the individual contributions of scientists who have discovered and developed drugs which save
and improve the lives of many people. The purpose of critical discussion is to
advance the understanding of the field. While many are spurred to criticize from competitive instincts,
"a discussion which you win but which fails to help ... clarify ... should be regarded as a sheer
loss." (Popper). Let us look at the present with the future in our mind. Although this article may seem
wide ranging and to some shows lack of focus, it is meant to be comprehensive and also show the lack of
focus of many solutions which attempt to solve the pipeline problem by revolving around the core problem.
The problems with drug discovery and development are already leading to international initiatives.
See also
Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products - USA,
Innovative Medicines Initiative (IMI) - Europe EU,
New Safe Medicines Faster Project - Europe EU
and the
Priority Medicines for Europe and the World Project "A Public Health Approach to Innovation" - WHO.
Drugs have both a humanitarian value and a financial value. Pharmaceutical research and development
contribute a major part of the research necessary to move new science from the laboratory to the bedside.
Through academic and industry efforts, many new drugs and devices have been developed and marketed,
which save and improve the lives of many people.
However, the costs to bring new drugs to the market have risen sharply in recent years and the output of drug development
and as such the Return On Investment (R.O.I.) has not kept pace. Drug development has always more or less
resembled more of a lottery than a controlled process, due to the lack of basic understanding of biological
complexity in the intrahuman ecosystem. In relation to the effort (cost involved), fewer drugs and biologics are making it
from Phase I clinical trials to the marketplace, which has dramatically increased the cost of drug development
(Crawford L.M., 2004). Late stage failure in
Phase III clinical trials and NDA disapproval has risen from 20% up to 50% (Crawford L.M., 2004).
From an economical perspective, the goal of improvements in drug discovery and development is to increase the
Net Present Value (NPV also
"fair value" or "time value") of pipeline molecules and to decrease the costs associated
with pursuing failed projects.
Basicaly the Net Present Value (NPV) is the worth of a good at the present moment and
for investments the Net Present Value is an important indicator. Only an investment, that offers
you a positive net present value, is considered to be worth to pursue. This has not been the
case in recent years for many drug development projects, as up to 90% fail.
The bottom line form an economical perspective is that in the end
any change in the process or its (scientific) content should improve the
Return on Capital Employed (ROCE).
This article aims at improving the probability of success in drug development (reduce late stage clinical development attrition)
by using better disease models (higher predictive power) in drug discovery an pre-clinical development. This improvement should
lead to bringing better drugs (more effective, less side effects) to the patients, both cheaper and faster.
The challenges which the pharmaceutical industry is facing:
From a business perspective, there are 2 sources of value creation by a more productive discovery processes
("clinical" quality of molecules). Both require a better scinetific engine in the first place:
There are 4 levers for creating value in (pre-)clinical drug development (process improvement):
What should we achieve for the overall drug development process in order to restore its productivity (principles):
What should be the deliverables (targets, quantitative), however ambitious they may seem given the present state of science:
The pharmaceutical industry has a history of initial innovative breakthroughs (first-in-class),
followed by slower, stepwise improvements of such initial successes (best-in-class).
How can we improve the Probability Of Success (POS) of the
overall drug discovery and development process and as such improve both the quality and
quantity of new drugs, both for innovative as well as stepwise improvements? Why do we need
to learn more about the human cytome to
improve drug discovery and development? How can cytome research help us to discover
and develop better drugs with a higher success rate in clinical development?
What is wrong with the drug discovery and development process as it is now, so its costs are
soaring and its R.O.I. is declining? Why do we only succeed in improving success rates somewhat with
biologicals and no longer with small molecules ?
Everyone
managing the discovery and development of drugs has to ask a few questions
about every new scientific idea or technology which pops up (and they do, all the time).
Every scientific idea or technology to be applied to drug discovery and development
must specify realistic and compatible goals and expectations. When we want to
introduce a new scientific idea into drug discovery and development we must
balance between good science and a credible business plan. We must be critical about the promises made.
Is the claim or argument relevant to the overall subject? ('Subject matter' relevance).
Is the argument or claim relevant to proving or disproving the conclusion at hand? ('Probative' relevance).
Personal note
My personal interest in cytomics, grew out of my own work on High Content Screening, as you can see in:
- Image Analysis gives a clear view in research
- Scalespace or Differential Geometry
- Color Differential Geometry or the Spatial Color Model
- Application of linear scale space and the spatial color model in light microscopy
- Automated Tiled Multi-mode Image Acquisition and Processing Applied to Pharmaceutical Research
- The M5 framework for exploring the cytome
Scope of the article
This article deals
with the analyis of several apects of the drug discovery and development process
and the the weak spots and flaws within this process, which cause the high late stage
attrition in the pipeline. The pharmaceutical industry has passed the threshold
where only slight adaptive changes can restore its productivity and profitability.
Reducing costs is not enough to restore the health of the industry, a paradigm shift is needed,
but this will require a new vision on the fundamentals of the drug creating process and the way true
knowledge and understanding is being built on the foundations of scientific discovery and through
applied research.
However important business processes are, this article in itself is not about
Business Process Improvement or Business Process
Reengineering as this is outside the scope of Research Process Improvement and
Research Content Improvement. The processes surrounding the drug discovery and
development process require attention and optimisation too, but in the end
success in drug discovery and development depends on bringing the right drug to
the right patient. Both the drug discovery and development process and its
scientific content require optimisation beyond their current state.
The
scientific content of the drug discovery and development process, goes beyond business management
principles and is more difficult to optimise than the process itself. It is the
present state of basic (reductionistic) and applied science and technology itself, in relation to the complexity of biological systems,
which still limits our chances for success. Inadequate understanding of basic science for certain diseases and the
identification of targets amenable to manipulation is one of the major causes of failure in drug development.
The endpoint of discovery is understanding a complex biological process, not just a pile of molecular data.
The endpoint of development is therapeutic success in man (a complex biological system), not just a molecular interaction.
The level of understanding at the end of drug discovery
(and preclinical development) should achieve a knowledge level which is capable
to predict success at the end of the pipeline much better than we do now.
No matter what is the origin of the
compound under evaluation, or how it came into being, a good description of its in vivo pharmacological properties
is necessary to assess its drug-like potential.
The sooner NCEs or NBEs evolve in a "rich" or lifelike biological environment
(resembling the situation in a human population) the sooner we capture (un-)wanted phenomena.
There is a time-shift between the implementation of a new
approach (linking genes almost directly to clinical diseases) and finding out
about its impact on commercial success, which makes the feedback loop
inefficient due to its long delay in relation to the quarterly and annual
business cycle. From a business perspective, any process can be sped up and
content can be sacrificed or complexity reduced. In a stage-gate process, the stages
deliver the content for the decisions at the gates, so the stages should be informative and
predictive. Processes and portfolio management can be optimised near perfection. This may be provide sufficient leverage
for a (albeit complex) 'nuts and bolts process' (e.g. automotive industry), but not for
processes in a biomedical context when our understanding of pathogenesis and
pathophysiology is still very patchy and
incomplete. We leave a large potential for improvement untapped. Improving
a development process which still fails for 90% of all developmental drugs,
is not optimised at all. With the current inefficient process we are, in most cases, unable to serve smaller patient populations.
If we ever want to reach the goal of personalized medicine, which is in my opinion is beter understood as
succeeding in unraveling the molecular diversity of clinicaly similar disease manifestations, some
conditions need to be fulfilled:
A lot of research and development will be needed to reach this goal, leaving aside the ethical minefield.
The complexity of
intermediary modulation of gene-disease (un-)coupling was clearly underestimated in recent years.
In the early stages of drug discovery, the data tend to be reasonably black and white.
As you get to more multifactorial information and more complex systems later on in drug discovery and development,
that becomes less true. Managing this complexity in a coherent way is a challenge we must deal with
in order to be successful. So how can we facilitate (and understand) the flow through the pipeline,
without generating empty downstream flow in clinical development? How to plug accelerators into the drug discovery and
(pre-)clinical development pipeline which prove their value onto the end of the pipeline? How
do we create a true Pipeline Flow Facilitation (PFF) process?
The first part of this article shows the problems of the drug discovery and development process.
It shows the present problems of the pharmaceutical industry.
The second part of this article looks for the best
way to improve the drug discovery and preclinical development process as these feed clinical development
with drug candidates which should make it to the right patients.
The third part of this article deals with the problems with
disease models in drug discovery and preclinical development and why they cause so much late stage
attrition later on in clinical development.
Overview of related articles on this website
Personal
interest and background where I provide som information how the idea for
a Human Cytome Project (HCP) has grown over time.
References
have been put together on one page.
Overview of problems and questions
Scientific background about
the Human Cytome Project idea can be found here
The potential impact on the
efficiency of drug discovery and development where I give an analysis
of the reasons for the unacceptable high attrition rates in drug development which
have now reached 9O%. Our preclinical disease models are failing, they look back
instead of forward towards the clinical disease process in man.
Overview of solutions and suggestions
A proposal of how to explore the human cytome where I give an overview of
the deliverables and the scientific methods which are (already) avalable.
How to deal with the analysis of the cytome in order to improve our understanding of
disease processes is being dealth with in another article. The
first part deals with the problems of
analyzing the cytome at the appropriate level of biological organization.
The second part deals with the ways of
exploring and analyzing the cytome at the multiple levels of biological organization.
A concept for a software
framework for exploring the human cytome is a high-level concept for large scale
exploration of space and time in cells and organisms.
Some thoughts on the pitfalls of applied research
The vulnerability of applied research, such as drug discovery and development, is hidden in the basics of scientific reasoning. In traditional Aristotelian logic,
deductive reasoning is inference in which the conclusion is of no
greater generality than the premises, as opposed to abductive and
inductive reasoning, where the
conclusion is of greater generality than the premises. Other theories of logic define deductive
reasoning as inference in which the conclusion is just as certain as the premises, as opposed to
inductive reasoning, where the conclusion can have less certainty than the premises. Scientific research is
to a large extent based on inductive reasoning and as such vulnerable to overenthusiastic generalizations and simplifications.
There is a lot more to say on the philosophy of science and its impact on research, but this is outside the scope of this article.
In addition the discussion about the problems of the drug discovery and development process is full of
red herrings and other
logical fallacies, which
distracts our attention from the real question: does the treatment work in man
(see also Organon from Aristotle).
Ignoratio elenchi
(also known as irrelevant conclusion) is the logical fallacy of
presenting an argument that may in itself be valid, but which proves or supports a
different proposition than the one it is purporting to prove or support (The promise
to the pharmaceutical industry "do this" or "buy that" and you
will deliver more and better drugs to the market). The ignoratio elenchi fallacy is
an argument that may well have relevant premises, but does not have a relevant conclusion.
The Red Herring fallacy is the counterpart of the ignoratio elenchi where the explicit
conclusion is relevant but the premises are not, because they actually support something else.
The complex relation between the input of
the drug discovery and development process (manpower, methodology and technology) and its output (drugs which succeed)
is underestimated, leading to unacceptable late stage attrition rates.
However, there are no simple answer to complex problems, such as how to create a truly productive process, both
effective and efficient.
The truth is forced upon us during the late stages of clinical development, when we fail because of a lack of
predictive power of discovery and preclinical development.
Like most opportunistic enterprises, pharmaceutical companies run the managerial risk of succumbing
to the enthusiastic optimism of a pragmatic fallacy.
Leading scientists and managers have to understand and systematically manage ambiguity in an
increasingly complex environment. There is more ambiguity in clinical reality and economical reality
than in an Eppendorf tube. You have to assess risk and benefits of decisions and anticipate
the impact on drug discovery and development in the longer term, far beyond the short-term quarterly goals.
Those who take responibility for strategic management have to grasp opportunities capable of
generating new opportunities for improving drug discovery and development in a productive way.
You have to scan the scientific, business and regulatory environment and think well ahead to identify things
which may get in the way of meeting objectives - either obstacles or changes in the overall situation.
Managers and scientists have to develop complex strategies which take into account the diverse interests across scientific domains,
economics, rules and regulations. It does not help that scientists prefer the scientific excitement of
reading Nature and Science, while
managers prefer the Wall Street Journal and the
Financial Times. There is a lack of cross-discipline
understanding and colaboration throughout the entire process (not enough silo busters). The true challenge is
to appreciate that the discovery, development, application, and regulation of the target to drugs pipeline
has to be viewed as integral processes with each element having important, sometimes critical,
implications on the other components with decisions weighed accordingly.
Drug discovery and development
Evolution of process and content performance
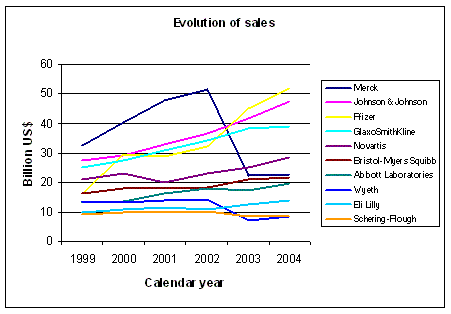
Figure 1: Evolution of sales for some big pharmaceutical companies. Source: Yahoo finance and other sources |
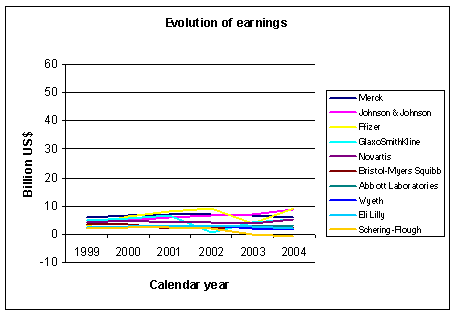
Figure 2: Evolution of earnings for some big pharmaceutical companies. Source: Yahoo finance and other sources |
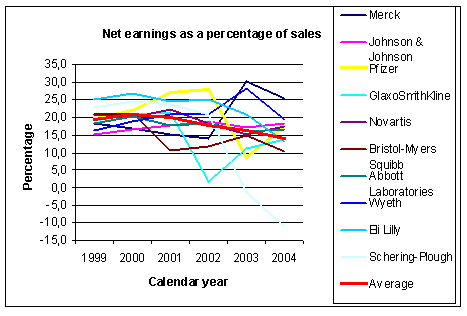
Figure 3: Evolution of earnings as ratio of sales for some big pharmaceutical companies. Source: Yahoo finance and other sources |
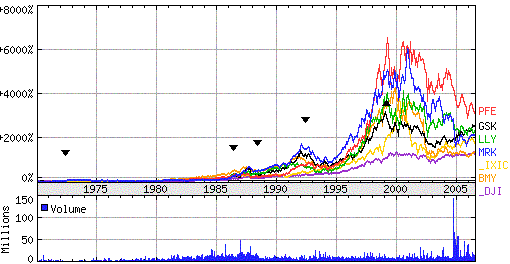
Figure 4: NYSE Pharma shares of Merck &Co. (NYSE:MRK), Pfizer (PFE), Eli Lilly (LLY), GlaxoSmithkline (GSK) and Bristol-Myers Squibb (BMY). _DJI (Dow Jones Ind.), _IXIC (Nasdaq). Source: Yahoo finance |
The graph in Figure 1 shows the evolution of sales for some big pharmaceutical
companies from 1999 until 2004. Pfizer, Johnson & Johnson and GSK show an increase ahead of the others.
Merck suffered from the problems associated with Vioxx, which illustrates the fragility of commercial
sucess. The graph in Figure 2 shows the evolution of net earnings from 1999 to 2004.
Here there is less difference between the companies, no company is able to outperform the others in a
dramatic way. The graph in Figure 3 shows the evolution of the percentage net earnings to
sales. On average there is a steady decline from 19.5% in 1999 down to 14.0% in 2004.
The graph in Figure 4 shows that in recent years the growth of the
pharmaceutical industry has slowed down. The pharmaceutical industry found themselves in a tight
spot in the beginning of the 21st century. The sector has seen a decrease in financial performance
following a boom period in the 1990s, fueled by a succession of drugs with sales over US$
1 billion per year (blockbusters). As all drug companies improved their stock value, the cause is common
to all of them and not to one company outperforming the other companies. Of all the knowledge required to develop
a new drug, the most important component is a true understanding of the molecular basis of the disease process.
This knowledge is mainly in the public domain and available for all companies, so no company is capable of
outperforming its peers in the long run. Although it may take up to 15 years to develop a new drug,
it may take up to 20-30 years to unravel the mechanism of a disease and this requires an effort on
a global scale, not just of a single pharmaceutical or biotech company. Almost 90% of all science required
to create a drug for a given disease, resides outside the pharmaceutical industry, but 90% of the risk is
hidden within the inefficient process used within the pharmaceutical industry (90% failure in drug development).
The industry faces the final challenge of proving that the ideas about a disease process truly work
in the complex biosystem of man.
The drug discovery and development process suffers from "particularism" and lack of "generalism"
as the success with one drug does not lead to a consistent increase of performance. Working on improving the process as it
exists today within the pharmaceutical and biotech industry is only leveraging 10% of the required knowledge-base.
A failure rate of 90% for 10 or 50 drugs in the development pipeline, is not an example of process improvement,
although the bigger pipeline will lead to a five-fold increase of drugs reaching the market.
How can the pharmaceutical industry
get out of the current situation of spiraling costs and reduced R.O.I?
There is no simple answer to this question, as a solution requires improvements in multiple domains.
The management of research must ensure that the resources are directed to investigations consistent with
the ultimate goal, the development of a successful drug. The management of
research is full of uncertainty and complexity. Research has substantial
elements of creativity and innovation and predicting the outcome of research in
full is therefore very difficult. The costs and risks involved in developing,
testing and bringing new drugs to market continue to grow, pharmaceutical
companies are coming under increased pressure to make the discovery and
development process more manageable and efficient. Today we need both better
processes as well as better science to succeed in the disease jungle or the
pathogen minefield. Success will go to those who can manage the hybrid
activities between science, technology, and the market.
Although innovative and
sound research is a prerequisite, it is ultimately the therapeutic success of the drug
which results in sales and profits. And it is usually proprietary (patented) products which earn
the highest returns, because they produce sustainable competitive advantage
over a substantial period of time (e.g. patent lifecycle). We must keep in mind
that it is the ability to produce proprietary products, not just interesting
science, which leads to a profitable and sustainable pharmaceutical company.
The business process around
the drug discovery and development process itself can be improved as well as
the R&D process management itself (e.g. Business Process Improvement,
process and portfolio management, ).
Reducing R&D costs and shortening product development cycles will certainly
contribute to an increase in profitability. But when the scientific substrate
of the R&D process itself is not optimised too, we leave a huge potential
for treating diseases cost-effectively and generating profit untapped.
Both the process and its content require our attention. The recent gulf of mergers
an acquisitions provides some short-term relief, but when we combine two companies
with each a 90% attrition rate in drug development, we just get a bigger
company with also a 90% attrition rate in drug development. This high attrition rate leaves
little margin for dramatic improvement of overall productivity. At the moment the
pharmaceutical industry is trying to generate some leverage by working on
improving the development process for the 10% developmental drugs which
make it through the pipeline. Business process engineering as such is less riskier
than rethinking the overall discovery and preclinical development process. Business
process improvement can be modeled on what was done in the automotive and aerospace industry
when those sectors faced hard times. Due to the success of its blockbusters in the 1990s, the
pharmaceutical industry only recently faced the same challenges.
I will focus on the content
of the drug discovery and development process. How can we scrutinize the
R&D projects earlier in the preclinical development process to help minimize
the risks involved in clinical development of new drugs (now down to 10% success rates)?
We need a better process content in relation to clinical reality, not only more content
as such.
Drug discovery and development:
an inefficient process
At the end of the drug
discovery and development pipeline, there are patients waiting for treatments,
company presidents and shareholders waiting for profit and governments trying
to balance their health care budget. For pharmaceutical and biotech companies,
the critical issue is to select new molecular entities (NME) for clinical
development that have a high success rate of moving through development to drug
approval. Finding new drugs (which can be patented to protect the enormous
investments involved) and at the same time reducing unwanted side effects is
vital for the industry. We must try to understand the reasons for failure in
clinical development in order to improve drug discovery and
preclinical development.
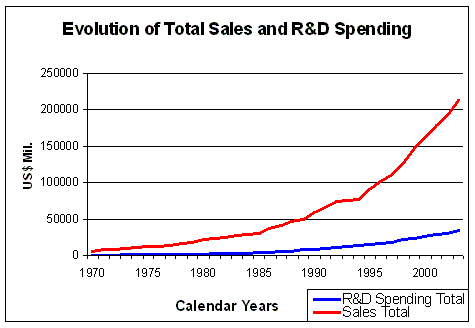
Figure 5. Evolution of Total Sales and R&D Spending Source: Pharmaceutical Research and Manufacturers of America (PhRMA) Pharmaceutical Industry Profile 2004 (Washington, DC: PhRMA, 2004) |
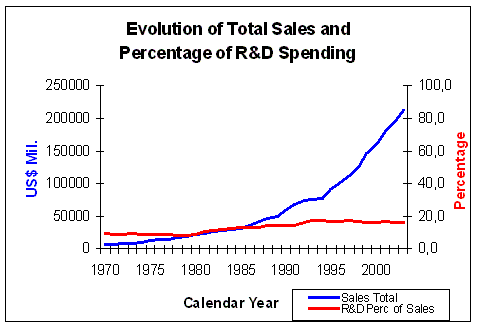
Figure 6. Evolution of Total Sales and percentage of R&D Spending Source: Pharmaceutical Research and Manufacturers of America (PhRMA) Pharmaceutical Industry Profile 2004 (Washington, DC: PhRMA, 2004) |
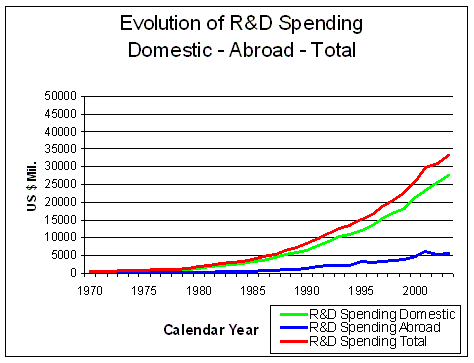
Figure 7. Evolution of Research and Development Spending Domestic and Abroad Source: Pharmaceutical Research and Manufacturers of America (PhRMA) Pharmaceutical Industry Profile 2004 (Washington, DC: PhRMA, 2004) |
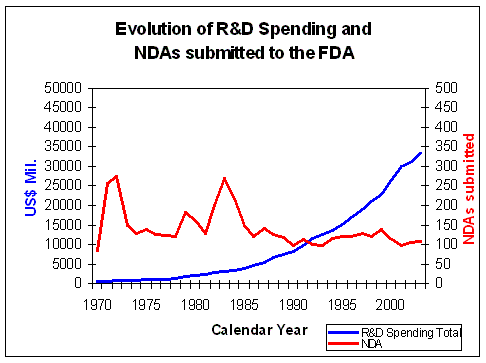
Figure 8. Evolution of Research and Development Spending and NDAs submitted Source: Pharmaceutical Research and Manufacturers of America (PhRMA) Pharmaceutical Industry Profile 2004 (Washington, DC: PhRMA, 2004) FDA CDER NDAs received per year |
The demand for innovative medical treatments is constantly growing as people in the
wealthy developed world live longer with a concomitant increase in the burden
of chronic diseases. At the same time, patient expectations about the quality
of treatment and care they receive are rising and unmet medical needs
remain high. There still are significant pharmaceutical gaps, that is, those diseases
of public health importance for which pharmaceutical treatments either
do not exist or are inadequate. What can modern society expect from its pharmaceutical industry to
deal with the challenges arising? Let us take a look at the evolution in income
of the US pharmaceutical industry and output over the last 30 years, from 1970 to 2003.
The total sales of the US pharmaceutical industry has risen almost exponentially
over the past 30 years (Figure 5). About 16% of sales income is spent
on R&D (Figure 6), which makes R&D, after marketing costs,
the second biggest item in the spending profile of large pharmaceutical companies.
The percentage of sale income spent on R&D has risen
from 9.3% in 1970 to 15.6% on 2003, a rise of 6.3%.
The total amount of money spent on R&D has risen
enormously since 1970, mostly in the US (Figure 7). In 2003, almost half of all R&D
spending worldwide was made in the USA.
However despite this almost exponential rise in total R&D
spending, the number of NDAs approved by the FDA has not risen significantly (Figure 8).
The money invested in R&D has not lead to an equal rise in output of the R&D process.
Due to the increasing mismatch between rising R&D expenditure and decreasing R&D
efficiency (Figure 8) the overall profit margins of the pharmaceutical industry
are decreasing (Figure 3).
Whatever the phrmaceutical industry spends on R&D, it has a significant overhead of additional manpower to sustain.
In 2000 the US pharmaceutical industry directly employed 247,000 people (down form 264,400 in 1993), with 51,588 of them
working in R&D, which means only 21% of its workforce is directly involved in the drug
discovery and development process (Kermani F., 2000; PhRMA 2002). In 2000 the European pharmaceutical industry
employed 560,000 people of which only 88,200 worked in R&D, which is 16%.
(EU source: The European Federation of Pharmaceutical Industries and Associations (EFPIA)
and The Institute for Employment Studies (UK)).
In 1990 the European pharmaceutical industry directly employed 500,762 people (76,287 in R&D or 15,2%). It took
10 years to increase total employment to 540,106 people (of which 87,834 in R&D or 16,3%, conflicting data),
but then it took only 3 years to increase total employment up to 586,748 (of which 99,337 in R&D or 16,9%).
The industry is not capable to reduce its overhead and to significantly increase its new drug generating workforce in
relation to its total employment. From 1990 to 2003 the European pharmaceutical industry icreased its workforce with 85,986
, but added only 23,050 for R&D.
The expenditures in R&D grow faster than its R&D workforce which indicates that
money is being spent mainly on equipment (e.g. for HTS), but which fails to sustain the growth of productivity in the
end (Figure 8). Ubiquity does not equal overall process efficiency and effectiveness.
Two elements which are often overlooked in the discussions about the increasing cost and duration
of R&D: tax returns and the
US Public Law 98-417
(the Hatch-Waxman Act) which was enacted in 1984.
Pharmaceutical companies in general spend a certain amount of the revenues on R&D because of its impact on
tax returns, so the cost is not the only driver. When sales increase tax deductions are important incentives to spend part
of the revenue on investments in R&D (Figure 6). But in the end, the new investments have to support
further growth, which is not always the case.
The "Drug Price Competition and Patent Term Restoration Act" (1984) was intended to balance two important public policy goals.
First, drug manufacturers need meaningful market protection incentives to encourage the
development of valuable new drugs. Second, once the statutory patent protection and
marketing exclusivity for these new drugs has expired, the public benefits from the
rapid availability of lower priced generic versions of the innovator drug
(Abbreviated New Drug Applications or ANDA).
One aspect of the "Drug Price Competition and Patent Term Restoration Act", the
"Patent Term Restoration"
refers to the 17 years of legal protection given a firm for each drug patent.
Some of that time allowance is used while the drug goes through the approval
process, so this law allows restoration of up to five years of lost patent time.
Under the
Hatch-Waxman Amendments,
patent protection can be extended (under certain conditions)
for up to 28 years, about 11 years of extra protection compared to the 17 years originally granted by US law.
The regulations governing the Patent Term Restoration program are located in the
Code of Federal Regulations (CFR),
Title 21 CFR Part 60.
The Uruguay Rounds Agreements Act (Public Law 103-465), which became effective on June 8, 1995,
changed the patent term in the United States. Before June 8, 1995, patents typically had 17
years of patent life from the date the patent was issued. Patents granted after the
June 8, 1995 date now have a 20-year patent life from the date of the first filing of
the patent application. Although pharmaceutical companies suffer from longer development cycles,
tax incentives and extended patent protection lessen the impact on their business results. The patients
are the true losers of the game, because they have to wait longer for new drugs for unmet medical needs.
Instead of creating a long-winded and inefficient process, medicine would be served better with a shorter and
more productive process.
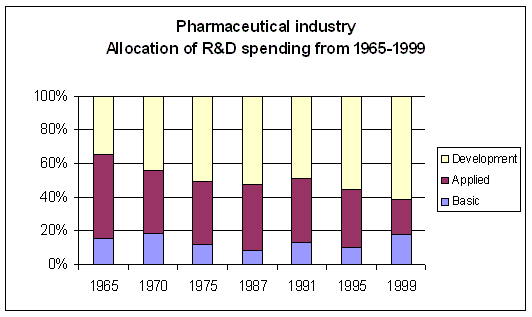
Figure 9. Evolution of R&D spending allocation Source: Pharmaceutical Research and Manufacturers of America (PhRMA) and Source: USA NSF Division of Science Resources Statistics (SRS) |
Although overall R&D spending has increased over the years, there has been a remarkable shift in the
allocation of R&D spending. Clinical development spending has increased significantly
(Figure 9), while spending on applied research (i.e. preclinical) has decreased.
Basic research spending shows an increase in recent years. The overall picture is an increased
spending on clinical development, while there is less spending on the processes feeding clinical
development with appropriate development candidates. Mainly the investments
in applied research, which is the bridge between basic research and clinical development shows
signs of neglect. The Early Development Candidates (EDC) were expected to require less preclinical
validation than before?
The cost to develop a single drug which reaches the market has increased tremendously in recent years
and only 3 out of 10 drugs which reached the market in the nineties generated
enough profit to pay for the investment (DiMasi, J., 1994; Grabowski H, 2002; DiMasi JA, 2003). This is mainly due to the low efficiency and
high failure rate of the drug discovery and development process.
Pharmaceutical companies are always trying
hard to reduce this failure rate. Indirect losses in drug development caused by
a failure in drug discovery are among the most difficult to quantify but also
among the most compelling in the riskmitigation category.
Pharmaceutical companies want to find ways to bring down the enormous costs
involved in drug discovery and development (Dickson M, 2004; Rawlins MD.,
2004).
Only about 1 out of 5,000
to 10,000 drugs makes it from early pre-clinical research to the market, which
is not an example of a highly efficient process. The focus of the
pharmaceutical industry on blockbuster drugs is a consequence of the mismatch
between the soaring costs and the profits required to keep the drug discovery
and development process going. The blockbuster model now delivers just 5%
return on investment and only one in six new drug prospects will deliver
returns above their cost of capital. The "nichebuster" is now an
emerging model for the post-blockbuster era.
Only diseases with patient
populations large enough (and wealthy enough) to pay back the costs for a full
blown drug development are now worth while working on. Research for new
antibacterial drugs is being abandoned, due to an insufficient return on
investment (R.O.I.) to pay for the development costs of new drugs (Lewis L,
1993; Projan SJ., 2003; Shlaes DM., 2003). If the industry cannot bring the
costs down, it may as well try to raise its income by changing its price
policy, but this shifts the solution for the problem from in- to outside the
company and places the burden on the national health care systems.
Companies which were more
successful in the past achieved a higher efficiency even without the
availability of extensive genomic and proteomic data and new low-level disease
models. The founder of Janssen Pharmaceutica,
Paul Janssen, PhD, MD (1926-2003),
in his early days achieved a ratio of 1 drug for every 3,000 molecules screened. Over the years he and his
teams developed about 80 drugs (out of 80,000 molecules, so 1 drug for 1,000 molecules screened)
of which 5 (6.3%) made it to the
WHO Model List of Essential Medicines.
He worked in fields as diverse as gastroenterology, psychiatry, neurology, mycology and
parasitology, anaesthesia and allergy. As a scientist he has been one of the most highly productive
and widely esteemed pharmacological researchers in the world for more than 45 years.
He had a deep understanding of both drug discovery and drug development. Dr. Paul Janssen
had always been the personification of a unique combination: on the one hand the
brilliant scientist, and on the other the very successful manager. Let us take a look at his approach to
active strategic management which
requires active information gathering and active problem solving. Dr. Paul Janssen practiced
Management By Walking Around (MBWA), which gave him access to all the research going on and
allowed him to orchestrate the efforts of his scientists, from discovery up to Phase III,
like a conductor and thereby avoiding silo development.
A deep understanding of a wide range of issues is required to bring a drug from early drug
discovery to the market. Introducing new technology and generating more data alone are not
sufficient to improve the drug discovery and development process (Drews J. 1999; Horrobin DF,
2003; Kubinyi H., 2003; Omta S.W.F., 1995). We need better content and understanding, not just
more targets and data to be fed into the preclinical and clinical development process. As such
the present-day discovery process, suffers from molecular
myopia as it lacks the big picture
understanding of disease mechanisms in man. In contrast the more traditional physiology based process, suffered from
system-wide
presbyopia as it lacked molecular resolution. The ideal approach would
be the combination of both, which has the potential to improve both the quantity as well as the quality of the process.
Quantity without a match in content quality (clinical relevance) leads to failure later on in the
drug development pipeline. We have to look at drug discovery
and preclinical development with clinical drug development and the patient in mind. Look back from
clinical reality into the drug discovery and development process and analyse its failures. A process which in the end
fails to prove its value in man should be changed.
The drug discovery and development process
Let us now take a closer look at the evolution of the output of the drug discovery and development over the years. How does the productivity of the process evolves? What is the cost/benefit ratio of the investments made and the overall outcome for drug discovery and development.
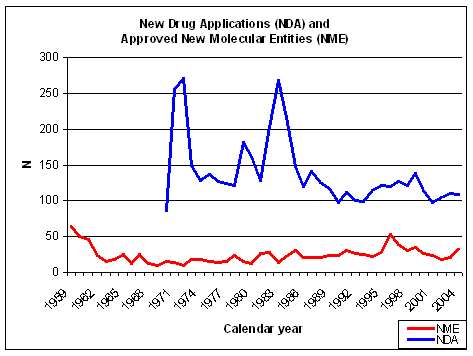
Figure 10. NDAs submitted over the years. Source: FDA CDER NDAs received and NMEs approved. |
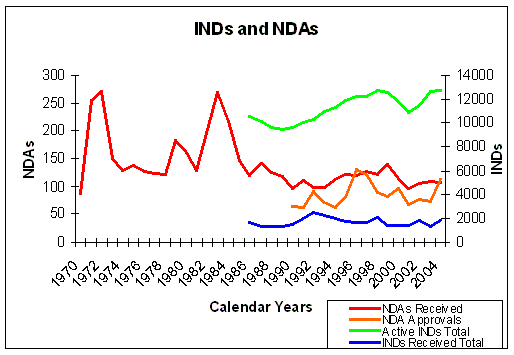
Figure 11. Evolution of INDs and NDAs over the years. NDAs left axis, INDs right axis. Source: FDA. |
The number of NDAs submitted does not show a significant increase in recent years (Figure 10),
compared to almost 20 and 30 years ago in the days of physiology based drug discovery. The number of
approved New Molecular Entities (NME) shows a sharp decrease in the early sixties, due to the more stringent
regulations for drug safety testing because of the
Thalidomide scandal. About 66% of the NMEs did not make it
anymore when better testing was required by the FDA and the pre-Thalidomide productivity was never reached again.
The number of NMEs was at its lowest at the end of
the sixties (9 in 1969) and has slowly increased since the early seventies (Figure 10). NMEs
are about 25% of all NDAs, before halfway the eighties it was on average less than 20%.
The number of NDAs is not the only
indicator of success for the pharmaceutical industry. Blockbusters generate higher sales per product, so
both the number of NDAs as well as the sales per marketed drug are important indicators.
Depending on a blockbuster makes a company vulnerable to problems (SAE) with a single drug and
patent expiry of a blockbuster has a bigger impact.
In figure 7 we can see that the number of INDs and NDAs submitted over the years, does not show a significant improvement.
The larger than average number of approvals in 1996 reflects the implementation of the
Prescription Drug User Fee Act (PDUFA).
The number of INDs does not show a significant increase over the years, so the overall productivity of drug
discovery and development has not improved, despite the high investments in Research and Development (Figure 7 and
Figure 11). The number of active INDs shows an overall increase, but this only means that
the drug development pipelines are filling up because the clinical trials take longer. The in- and
outflow of drug development (INDs and NDAs) has not changed in a way to explain the increase in active INDs.
The pharmaceutical industry itself expects that products will stay in phases longer than has historically
been the case, lowering the probability of a product moving from one phase to another in a particular year.
We have not seen a proportional increase in NDA submissions to the FDA, compared to the number of active INDs
(Figure 10 and 11).
The main reasons for declining productivity of drug development are:
- Tackling diseases with complex etiologies, which are not well understood.
- Demands for safety, tolerability are much higher than before.
- The proliferation of targets is diluting focus.
- Genomics has been slow to influence day-to-day drug discovery.
- A negative impact of mergers on R&D performance?
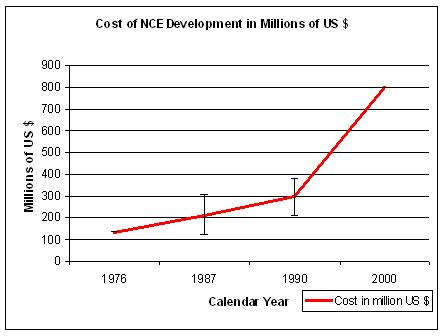
Figure 12. Sources: for 1976 Hansen, 1979; for 1987a Wiggins, 1987; for 1987b Woltman, 1987c, for 1987c DiMasi, 1991; for 1990a and b OTA pre-tax, for 2000 DiMasi, 2003. Differences are also due to out-of-pocket versus capitalized costs. |
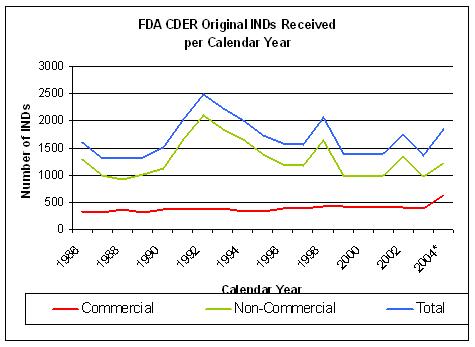
Figure 13. Source: FDA CDER INDs received per year |
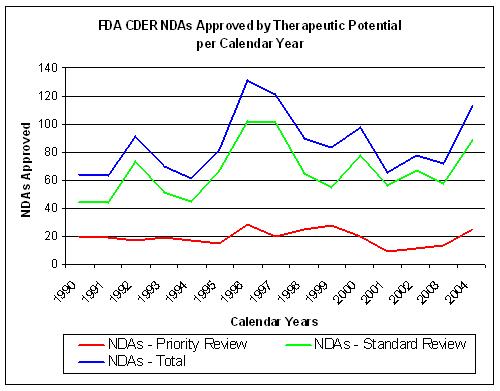
Figure 14. Source: FDA CDER NDAs approved per year |
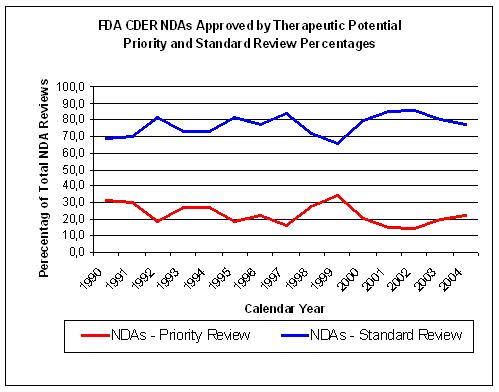
Figure 15. Source: FDA CDER NDAs approved per year |
Let us now take a closer look at the drug discovery and development process
(clinical trials).
Although different sources give different outcomes, the trend is one of increasing costs and reduced Return On Investment (R.O.I.).
In 2000 it took about US$ 500 to US$ 802 million to develop a new drug and bring it to the market (DiMasi J.A., 2003),
which is a significant rise since 1976 when it cost about US$ 137 million (all numbers in year 2000 US$) (Figure 12).
These estimates include opportunity costs, which are lost profits that could have been realized if the money tied
up in an enterprise had been invested elsewhere (DiMasi J.A., 2003). Almost half of the
DiMasi (Tufts) US$ 802-million
figure - $399 million - is comprised of this "cost of capital", leaving a figure of US$ 403 million for
direct out-of-pocket expenses, most of which is expended in clinical trials. Whether you favor US$ 403
million or US$ 802 million, the cost of drug discovery and development is far too high.
When we look into the diferent stages of drug discovery and development,
the US$ 802M costs are divided over: Discovery and preclinical testing US$ 335M, Phase I: US$ 141.7M,
Phase II: US$ 137.2M and Phase III: US$ 174M. The total cost for clinical development is
US$ 452.9M. The cost for FDA Review/Approval: US$ 13.8M.
The basic numbers for time
spent and costs made in drug discovery and development can be found in several
documents published by institutes which generate reports about the
pharmaceutical industry (Boston Consulting Group,
Tufts Center for the Study of Drug Development,
Pharmaceutical Research and Manufacturers of
America (PhRMA), the Institute for Regulatory Science (RSI),
CMR International, etc.).
IMS is a source for pharmaceutical market information. The
Association of Clinical Research Professionals (ACRP)
and the Center for Information and Study on Clinical Research
Participation (CISCRP) provide information on clinical trials.
To be complete, there are
alternative views
which criticize the calculation of the cost of drug discovery and development.
Here are the
Public Citizen and
TB Alliance reports.
A discussion of these reports and the Tufts study can be found
here.
Although an open and critical discussion is the only way to understand complex
issues such as research and development costs, the discussion sometimes loses its focus and becomes tainted by
sophisms
to support political and personal agendas. I leave it to the critical reader to decide.
The consequence of accepting the alternative views would be that the
pharmaceutical industry would be losing money due to costs outside its core
mission, which is even worse, because research and development can be improved,
but this would not help in this case. The result is in each case, that drugs
are only worth while to develop, if they have an enormous market potential
(large numbers of wealthy patients),
require as little as possible investments (me-too drugs and generics)
and have the shortest development cycle possible (less complex diseases).
Otherwise they do not earn back the money invested, when finally they
reach the market. This leads to an increasing focus on typical Western "diseases"
such as obesity or hypercholesterolemia, due to overintake of food and
unhealthy living. Tropical diseases, if they do not make it to the wealthy
world, are to be avoided. You cannot blame the pharmaceutical industry,
because if they do not live up to the expectations of their shareholders, they
are punished by a decreasing stock-value (see Figure 1).
The number of INDs coming out of drug discovery does not show a significant improvement since 1992
although overall costs have risen sharply.(Figure 13 and 14).
The non-inovative drugs get a standard review by the FDA instead of a priority review and
constitute about 75% of all NDA submissions (Figure 15).
About 10-20 % of the total costs are due to the drug discovery process, the rest is
caused by drug development, production and marketing costs. Clinical development
costs, on average US$ 467 million, which makes up more than half the total cost.
The cost of a Phase I clinical trial is about US$ 15.2, for Phase II it costs about US$ 16.7 and
Phase III US$ 27.1 (in 2000 US$, DiMasi J.A., 2003). The cost of a Phase III clinical
trial ranges between US$ 4 million and US$ 20 million and you need at least two of
them (Kittredge C, 2005).
Study delays, such as slow patient recruitment, protocol amendments and review
processes, are contributing factors. Every day that a drug is prevented from being
on the market means a loss of sales, which in the case of blockbuster drugs can be
as much as US$ 45 million per day.
From about 8 years in the
1960s it now takes an average pharmaceutical company about 10 to 15 years to
bring one new drug to the market. Of these 15 years about 6.5 years or 43%
of the total time is spent in pre-clinical research.
Development starts with candidate/target selection or the selection of a promising
compound for development. Pre-clinical and
non-clinical research involves necessary animal and bench testing before
administration to humans plus start of tests which run concurrently with
exposure to humans (e.g. two-year rodent carcinogenicity tests).
About 7 years or 46 % of the total time is time spent in clinical research (1.5 years
in Phase I, 2 years in Phase II and 3 years in Phase III). Phase I (First Time In Man, FTIM) of a clinical trial
deals with drug safety and blood levels in healthy volunteers (pharmacology). Phase II (Proof of concept, PoC) deals
with basic efficacy of a new drug, which proves that it has a therapeutic value
in man (exploratory therapeutic). Finally Phase III deals with the efficacy of the drug in large patient
populations (confirmatory therapeutic). It is easy to understand that the increase of the population used
to study the effect has a dramatic impact on the complexity and the cost of the
clinical trial.
To process a New Drug
Application (NDA) takes the U.S. Food and Drug
Administration (FDA) on average 1.5 years based on the results and
documents provided by the pharmaceutical industry. The situation in
In the 1990s about 38 % of the drugs which
came out of discovery research dropped out in phase I. Of those molecules which
made it out of phase I, 60 % of those failed in phase II clinical
studies. And now we get to the really expensive phase III in which 40 %
of the remaining candidates failed. Of those drugs which made it out of phase
III to the FDA 23 % of the ones that made it through the clinical trials
failed to be approved by the FDA. All this translates to about 11 %
overall success rates from starting the clinical trials (
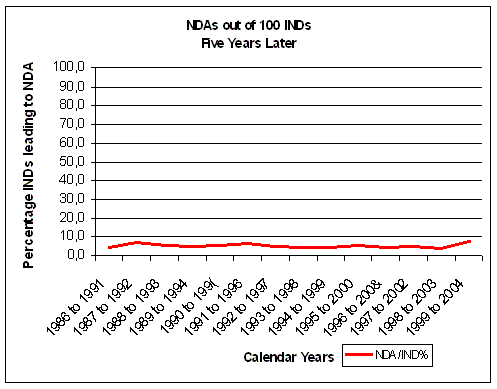
Figure 16. Less than 10% of INDs make it to an NDA. Source: FDA CDER NDAs approved per year and FDA CDER INDs received per year |
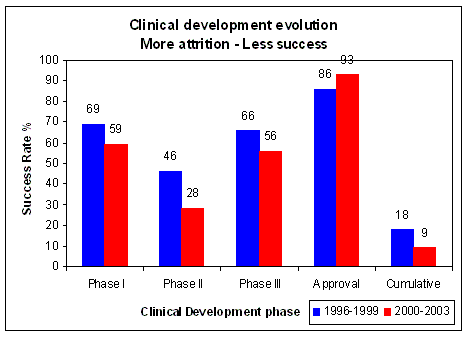
Figure 17. Overall success of clinical development decreased from 18% to 9%, worst decline in Phase II (effectiveness), from 46% to 28% Source: Loew C.J., PhRMA, HHS Public Meeting, November 8, 2004 |
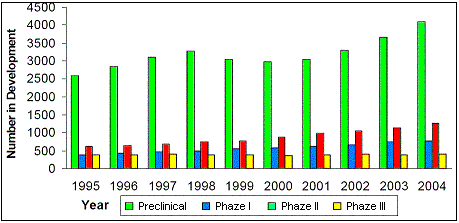
Figure 18. Evolution of attrition from 1995 to 2004. Source: Pharmaceutical Research and Manufacturers of America (PhRMA) |
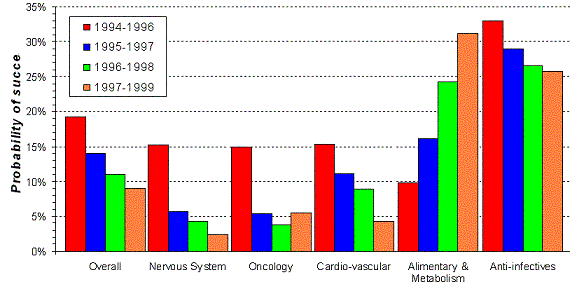
Figure 19. Trends in probability of success from 'first human dose' to market by therapeutic area. Source: Pharmaceutical Research and Manufacturers of America (PhRMA) |
As a rough indication of overall inefficiency we
can compare FDA NDA and IND data five years different. If we take on average 5 years from IND (IMP in Europe)
after 5 years of IND filing, less than 10% of INDs make
it to an NDA (Figure 16). The evolution of NDA approvals also shows a decline over the years.
In recent years overall success rates for clinical development decreased from 18% to 9% (Figure 17).
This is mainly due to an almost 40% reduction of success in phase II clinical trials, which means a failure in
exploratory treatment or clinical activity. A Phase II clinical trial is intended to determine activity, it does not yet
determine efficacy, which is the goal of a Phase III clinical trial. Thus the outcome of Phase II is a decisive
point in a drug's development. If we look at the evolution of attrition rates from 1995 to 2004, we see an
overall increase in development candidates in preclinical development and an increase in Phase I and II development
(Figure 18). There is no significant increase in Phase III clinical trials, as most developmental
drugs increasingly fail in Phase II. The drugs show an activity in drug discovery and preclinical development,
but no significant activity in a clinical situation on a real-life disease process. The increase in attrition
is not the same for every therapeutic area (Figure 19). For alimentary and metabolic diseases
the probability of success (POS) is even increasing and is about then times as high as for the nervous system (1999).
The high success rate of anti-infectives is also caused by the fact that we are capable to make a valid representation of
the entire target system (bacteria) in a "test-tube" or "petri-dish" early on in the process,
and not only a billion-fold reduced representation of the human biosystem. As long as the dominant view on
applied science remains that a set of molecules in a "test-tube" can represent the complexity of
system (reductionism) our models will disappoint us at the end of the process when there is no
escape from the complexity and variability of man and human population. Leaving too much of the original
system out of an experiment brings too much flaws into the experiment. Elimination without confirmation
of validity against the original condition gives flawed results (and late stage attrition).
The significance of increasing Phase II failures is a new evolution, as in the
1980s and early 1990s the failure rates remained relatively steady. The failure rate of new clinical
entities (NCEs) remained relatively steady through the 1980s and early 1990s (DiMasi J.A., 2001).
Among NCEs for which an investigational new drug (IND) application was filed in 19811983,
approval success rates were 23.2%; 19841986, 20.5%; 19871989, 22.2%; and
19901992, 17.2%. This includes both self-originated and acquired NCEs. According to the FDA
historically 14% of drugs that entered Phase I clinical trials eventually won approval, now 8% of
these drugs make it to the marketplace, and that half of products fail in the late stage
of Phase III trials, compared to one in five in the past
(Crawford L.M., 2004).
"...In the past, we used to see a 20 % product failure in the late stages of the Phase 3 trials.
Currently, the failure ratio at this stage is 50 %. The reason for this unpredictability,
in our analysis, is the growing disconnect between the dramatically advancing basic sciences
that accelerate the drug discovery process, and the lagging applied sciences that guide the
drug development along the critical path. ..."
(Crawford L.M., 2004). Overall
late stage attrition is on the rise, but how should preclinical development and Phase I clinical trials
predict success or failure in Phase II or III, when they are not conceived or designed to do this?
Each stage from discovery over preclinical development to clinical development is meant to provide
an answer for a particular question, not for the question arising in the next stage of the discovery
and development pipeline. Which elements or markers in preclinical development would allow us to
predict events in clinical development. In order to achieve this we need a better understanding of the critical
issues in the clinical disease process. The analysis of failures in Phase II should at least help us
to understand the mechanism of these failures in order to feed those lessons back into preclinical
development. The transition from preclinical development to Phase I and Phase I itself deals with finding a
appropriate dosing scheme to start with (e.g. MTD Maximum Tolerated Dose), but not yet with clinical
activity, which comes into play at Phase II.
There are some practical considerations to determine the clinical activity of a developmental drug,
one of which is the sample size. The study design (case/control, cohort study, RCT, etc.) is the
first decision, but sample size is a close second.
An important issue is the power of the trial. Once the level of activity that is of interest has been decided
on, one should design a trial that exposes the fewest possible patients to inactive therapy,
e.g. by appplying the method of Gehan and Schneiderman (Gehan E.A., 1990). In general you need more patients
when you want to find out about a smaller therapeutic effect. This is an important cause of the
overall increase in patient numbers required, depending on what you want to prove. When we cannot achieve a
dramatic therapeutic breakthrough with a diseases, a small improvment is what we want to prove. Instead of a
revolutionary breakthrough, quite often therapeutic improvements are only incremental. Let me clarify this with an example.
When Louis Pasteur (1822 1895)
developed a vaccine against rabies, the shortterm outcome was clear, either you died or you survived. Rabies is
a viral disease with about 100% mortality, i.e. you almost always die when you get the disease.
So the therapeutic effect was very simple to assess, which also made complicated analysis of the therapeutic
results less necessary. There was also less consideration about possible side effects, as dying from rabies was
a horrible disease process.
Let us now take a look at Alzheimer's Disease (AD) (named after
Alois Alzheimer), a debilitating degenerative disease
of which the pathological process is still not well understood. We cannot achieve a "restitutio in integrum"
(restoration to original condition) and regrow the brain cells which are lost due to the disease process.
So, now we can decide to wait until we know all about the process and then start developing a cure. This would mean that
in the mean time we do nothing to help whatsoever. As you can understand, this is not a valid option.
In the mean time, therapy is aimed at slowing down the process of mental deterioration. This however is a
more subtle outcome than the short-term live or die outcome in the case of rabies. These less than 100% success
rates make it harder to prove the success of a new therapy. The need to prove a small improvement, makes clinical trials
more complex and much larger.
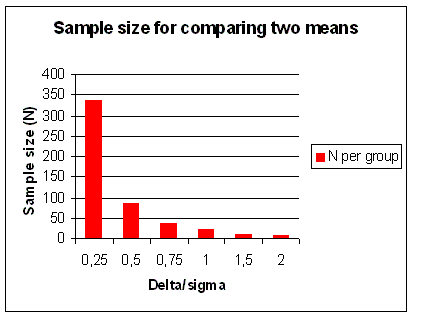
Figure 20. Sample size (N) for comparing two means. In addition to α and β, N only depends on Δ/σ, or the effect size. α = 0.05 and 1 - β = power = 90% for a 2-sided test. The graph shows N as a function of Δ/σ = difference in units of s.d. |
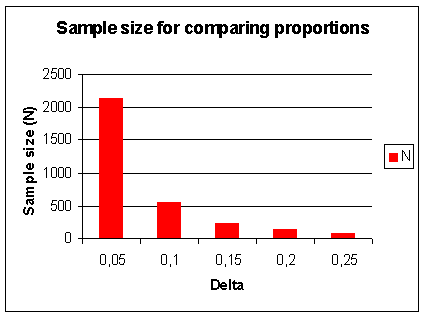
Figure 21. Sample size (N) for comparing proportions (p). In addition to α and β, N depends on p and Δ. Let α=0.05, 1- β = power = 90%, 2-sided testing, p=0.5 (conservative estimate for variance). The graph shows N as a function of Δ = difference p1-p2, e.g. 0.2 = 0.6 - 0.4 |
Designing a clinical trial is not a trivial endeavour as the days
of Louis Pasteur are gone and the environment in which to develop new therapies has changed dramaticaly. A clinical trial
requires careful design in order to be able to answer the research question (hypothesis) with some confidence in the answer.
You want to prove that a new therapy works in a reliable way. Traditionally, H0 is the hypothesis that includes equality or the
expectation that nothing will happen and the alternative hypothesis H1 that something significant will happen (Rosner B, 1995).
A p-value is a measure of how much evidence we have against the null hypotheses. The significance test yields a p-value that
gives the likelihood of the study effect, given that the null hypothesis is true. A small p-value provides evidence against
the null hypothesis, because data have been observed that would be unlikely if the null hypothesis were correct.
Thus we reject the null hypothesis when the p-value is sufficiently small. However life in clinical trilas is not that simple.
There are two type of statistical errors you can make in a trial. A Type I error occurs
when you reject H0 when H0 is true, i.e., you declare a significant difference when the result happened by chance
(false positive - a drug will be used while it is not effective). A Type II error occurs when you accept H0 when H1 is true, i.e.,
you say there is no significant difference when there really is a difference (false negative - a drug will not be used while it has an effect).
How do we deal with these issues? While we cant prevent the possibility of incorrect decisions, we can try to
minimize their probabilities. We will refer to alpha (α) and beta (β) as the probabilities of Type I and
Type II errors, respectively.
or
An interesting element of a trial is the power of the trial. A study can have too little power to find a meaningful difference,
when the sample size is too small. No significant difference is found and the treatment or method is discarded when
it may in fact be useful. The alternative Hypothesis (H1 or Ha) is that there will be a significant (therapeutic) effect.
The P(Type II error) = β and β depends on how large the effect really is. The power (P) of a test is the probability that we
reject the null hypothesis given a particular alternative hypothesis is true and Power = 1 - β. Summarized:
β = Probability(missing the difference) and Power = Probability(detecting the difference).
All this comes down to the overall rule that in order to prove a small decrease in disease progression we need
a relatively large number of a patients. It is because of this kind of effect, the size
of patients in clinical trials has risen dramaticaly in recent years. Also in the case of rabies, there was no
effective treatment to compare with, so the comparison was straightforward and simple. In Figure 20 and
Figure 21 you can see for two different types of trials, the effect of sample size required to detect an increasing
difference. This is an important reason for having patient populations of up to 5,000 patients in Phase III clinical trials.
If we could make a big difference with a treatment, then we would not need such large numbers of patients to prove our case.
With a chronic degenerative disease, reducing the speed of progress of the disease with only 0.1%, could
mean that in 20 years thousands of people would benefit (longevity in the Western world), but the problem is that you must
prove this small difference within the scope of a clinical trial. This is one of the most important reasons for clinical trials
to become increasingly global in nature and more complex in protocol design.
The difference with the 19th century is also that we now have to compare with drugs which are already
on the market and have a proven therapeutic effect. The pharmaceutical industry is increasingly challenging itself to improve
against its own therapeutic success of the past. As such the pharmaceutical industry itself is the biggest problem for the
pharmaceutical industry. There is a lot more to be told on on clinical trial design, but this
is not within the scope of this article. The main issue is that in modern clinical development, the situation is more
complex to evaluate than before.
In inductive research, applying statistics has to be done with care. Expanding a trial population beyond
the boundaries of statistical relevance, may lead to spurious statistical significance but will not improve
the correlation to clinical relevance. By doing this we increasingly feed the process with false positives
and increase the pressure towards the end of the pipeline. The basic principles of probability (significance)
and induction (relevance) should be taken into account when designing and performing experiments.
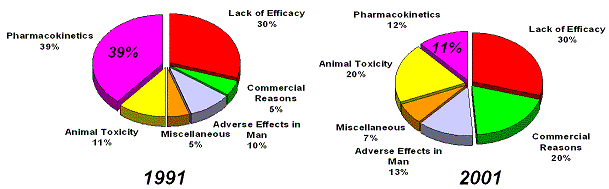
Figure 22. Despite a reduction in attrition due to pharmacokinetics issues, efficacy has not improved, since 1991. In 1991 40% of PK failures were caused by poorly bioavailable anti-infectives, when we remove these from the equation, then only 7% of failures in 1991 were caused by poor ADME. Source: Pharmaceutical industry attrition profiles, evolution (Kennedy, T., 1997; Prentis RA, 1988). |
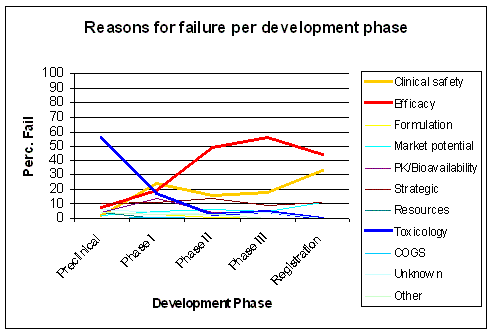
Figure 23. The major cause for failure, efficacy, only becomes apparent late in development. Source: KMR Group 1998 - 2000 |
What about the evolution of the basic reasons for attrition in drug development? Attrition due to a lack of efficacy
of drugs in development has not improved since 1991 (Kennedy, T., 1997; Prentis RA, 1988) (Figure 22).
Attrition rates due to poor pharmacokinetical profiles (PK) have dropped significantly, due to better preclinical
in-vitro and in-vivo models. However about 40% of failures in clinical development were due to inappropriate
pharmacokinetics of poorly bioavailable anti-infectives, if those were removed from the equation
then ADME was only responsible for 7% of failures in 1991 (Kennedy, T., 1997).
The basic numbers on attrition causes explain why attrition rates in Phase I clinical trials have declined less
than those in Phase II. Drugs with unfavorable PK profiles are now increasingly stopped before they reach
clinical development, so the ineffective ones now make it into Phase II in relatively larger numbers.
The clinical development attrition trends also show an unfavorable evolution since 1991 (Figure 22).
The disease models used in drug discovery and preclinical development fail to predict
failure in clinical development in about 80 to 90% of the drugs which enter clinical development.
And the combined predictive power of all clinical trials (Phase I to III) fails to predict failure in 1 out of
four or 25% or even 50% of all drugs submitted to the FDA for approval.
The major cause of attrition, efficacy, also shows up late in development, as preclinical development and
Phase I are unable to detect this failure. Preclinical development lacks the proper predictive models
and Phase I is not designed to detect a failure in efficacy. Clinical safety issues increase with
the number of people taking the drug, after it is on the market (Figure 23).
What can we learn out this
numbers and what is being done in drug discovery? The role of
absorption,
distribution, metabolism, excretion (ADME) and toxicity (ADMET) is an important part
of the drug discovery process as ADMET is an important cause of failure in drug
development (Yan Z, 2001; Lin J, 2003; Nassar AE,
2004). Pharmaceutical profiling assays provide an early assessment of drug-like
properties, such as solubility, permeability, metabolism, stability and drug-drug interactions (Di L., 2005).
The drug discovery process (target identification, target validation,
lead identification/optimization ) and preclinical development such as ADMET
studies, fail to predict the failure of a drug in clinical development for 4
out of 5 or at least 80 % of the molecules which enter phase I. The rates
of failure in expensive Phase III trials in oncology are the worst in the
industry (Kamb A., 2005). Improving the predictive power
of disease models in drug discovery, preclinical development and ADMET is an important issue to reduce
the late stage attrition rate in drug development.
A new drug spends about 90
% or 13.5 years of his career within the discovery and development
process, before it reaches the FDA for the last 10 % or 1.5 years. So the
FDA does not account for the majority of the time it takes to bring a new drug
to the market, nor does it account for the majority of failures which is only
20-25 % or 1 out of 5 or 1 out of 4 drugs which enter phase I or 1 out of
5,000 (0.02 %) if we start from the beginning of the process. Although
the investments in the early stages of the drug discovery process have
increased tremendously, this means nothing compared to the cost of failure in phase
III of a clinical trial.
The manufacturing process
After the drug discovery
and development is finished for a particular drug, the
drug enters the market and is being manufactured. Making manufacturing more
efficient is also an imperative for the pharmaceutical industry. The 16 largest
drug companies spend more than twice as much on manufacturing as they do on
R&D, according to a recent study by GlaxoSmithKline,
Companies are under increased regulatory pressure for manufacturing, such as the Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients (ICH Q7A), FDA Good Manufacturing Practice (GMP) and product labeling. The Good Automated Manufacturing Practice (GAMP) organization was founded in 1991 by pharmaceutical experts to meet the evolving FDA expectations for GMP compliance of manufacturing and related systems. Impending requirements being imposed by the FDA in the U.S. and the EMEA in Europe require companies to submit product labeling content in highly structured XML formats (Structured Product Labeling (SPL) in the US and Product Information Management (PIM) in Europe). Distribution of drugs is regulated by the Good Distribution Practice (GDP) of Medicinal Products for Human Use. New initiatives are being taken to improve the overall manufacturing process. Process Analytical Technology (PAT) provides a framework for innovative pharmaceutical manufacturing, control and product quality assurance. FDA Process Analytical Technology Initiative (PAT). The EUFEPS Process Analytical Technology Sciences.
The FDA wants to deal with the growing public health problem of counterfeit prescription drugs in the United States. Counterfeit drugs are not only illegal but are also inherently unsafe. A famous case of the withdrawal of a drug due to deliberate product tampering was the Tylenol murder case. The Tylenol murders occurred in the autumn of 1982, when seven people in the Chicago, Illinois area in the United States died after ingesting Extra Strength Tylenol medicine capsules which had been laced with cyanide poison. This incident was the first known case of death caused by deliberate product tampering. Johnson & Johnson was praised by the media at the time for its handling of the incident, although it cost the company about US$ 100M in lost revenues (see also Johnson & Johnson Credo). In the near future the FDA will require that the industry to implement full-scale RFID serialization (needed for closed-loop drug tracking) and electronic pedigree (ePedigree) applications (needed to find and prosecute violators) The Radiofrequency Identification Technology (RFID) is meant to monitor and protect the U.S. drug dupply chain. Radio Frequency IDentification (RFID) is an automatic identification method, relying on storing and remotely retrieving data using devices called RFID tags or transponders. In general, authentication systems that operate independently from the underlying data collection technology will help the drug industry secure the drug supply, protect valuable brands, and avoid legislation that will force costly compliance requirements that add little business value.
Inspection by the FDA are not to be taken lightly. Pharmaceutical, Medical Device, Biopharmaceutical and Generic Drug companies all face a common dread. The FDA has called, and they are coming to audit their manufacturing facility. At the "FDA's Electronic Freedom of Information Reading Room", the FDA publishes the findings of its inspections on-line: Warning Letters and Responses.
Vulnerability in Phase IV
The pharmaceutical industry depends on a relatively small number of active components.
While there are around 10,300 FDA-approved drugs in the United States today, most of these are made up
of some combination of only 433 distinct molecules. Half of these 433 molecules were approved before 1938,
and at least 50 are "me too" drugs, a slightly modified form of a compound already on the market. Finally,
there are only eight major, chemical "scaffolds" upon which all the 433 molecules are based.
Due to the difficulty and inefficiency of the drug discovery and development process, pharmaceutical
companies rely on only a few drugs for their income and profit. This makes them extremely vulnerable
for massive income loss when one of the drugs encounters problems after it is on the market.
Serious problems with a drug after it has been on the market in general means lawsuits against the
company and a serious blow to its reputation (e.g. pharmacovigilance or Phase IV trial). Each year about 17,200
Adverse Events (AE)
and 800 Serious Adverse Events (SAE)
are typically reported to the FDA for newly approved drugs (Source: FDA).
Seven of the 303 (2.3%) new molecular entities (NME) approved by the FDA between January
1994 and April 2004 were withdrawn from the market due to safety concerns. Although 97.7%
of NMEs do not cause such safety problems, the 2.3% which do, bring the pharmaceutical
industry in trouble. Older drugs can also be a major cause of hospital
admissions, such as with aspirin (Pirmohamed M, 2004). The perception that new drugs are less safe
than older ones is not always true.
However the accompanying harm to patients and the billions spent developing
and marketing the drugs are a big problem for the industry.
No amount of testing can guarantee to find all of the possible side-effects for every person
who may take a medicine. A reaction which occurs at a rate of 1 in 100,000 people or
even at a higher rate of 1 in 10,000 for instance, may not be seen until very large numbers
of people use the medicine. Even the largest clinical trials are underpowered to detect rare events
before a drug hits the human population at large. The increasing attention to chronic diseases for which there is no
"restitutio ad integrum" possible, but only long-term treatment also increases the
exposure of individual patients to the drug, which we cannot foresee during clinical trials before the
drug hits the market. Even with these odds, no pharmaceutical company
wants to be in the news with Serious Adverse Events (SAE)
about a drug already on the market. Being the CEO of a pharmaceutical company is not something for
the faint of heart. One day a company is praised for a new breakthrough drug, the next day
it has its name in the news associated with lethal side effects of another drug. Some recent
events have shown that pharmacovigilance principles and procedures are in need for improvement.
The EU European Risk Management Strategy (ERMS) of 2002 is an example of such an initiative. It aims at
strengthening the EU Pharmacovigilance System (see also EudraVigilance).
Current methods in pharmacovigilance often use monitoring and simple analysis of safety signals after they have been detected in the postmarketing process. Sometimes Phase IV clinical trials (postmarketing) reveal important side effects which were not discovered before. This was the case for Vioxx according to the APPROVe study by Merck. The cost of missing a safety signal or not detecting it before it affects the general population is huge. The withdrawal of a drug from the market has serious consequences both due to the loss in revenue for the company and the financial consequences of lawsuits. As was the case with Vioxx, a pharmaceutical company may turn to unethical and unlawful pressure on scientists and doctors to protect its commercial interests (Moynihan R., 2009; Cahana A, 2006). Manipulating opinion about a drug as was the case with Vioxx in the ADVANTAGE seeding trial of Merck and guest authorship and ghostwriting is not unusual in the pharmaceutical industry (Hill KP, 2008; Ross JS, 2008).
The cost of an adverse drug reaction on an average per patient basis is about € 2800 (approximately US$ 3,360) in hospitalization costs alone (Gautier, 2003). The total losses to a company can reach billions of dollars from the loss of reputation and revenue and from medical and litigation expenses.
Some examples of
Serious Adverse Events (SAE) over the years give an indication of the impact on the lives of people,
society and the pharmaceutical industry. An inadvertently toxic preparation of sulfanilamide
had a central influence on the US Food and Drug Administration (FDA). A preparation called
"Elixir Sulfanilamide" contained diethylene
glycol as a solvent, which is toxic. This preparation killed over one hundred people,
mostly children, and led to the passage of the 1938 Food, Drug, and Cosmetic Act
(the 1937 Elixir Sulfanilamide Incident).
Thalidomide (Softenon) was withdrawn from the
market in the sixties when thousands of babies were born with deformities as a
result of their mothers taking Thalidomide during pregnancy (McBride WG, 1961). Thalidomide never
made it to the USA in the sixties, mainly due to
Dr. Frances Oldham Kelsey
of the FDA, who refused to authorize thalidomide for market when she had serious concerns about the drug's safety.
In the USA the Thalidomide case lead to the Kefauver-Harris Drug Amendments (1962) to be applied retroactively to the
Federal Food, Drug, and Cosmetic Act (1938). In Europe the Thalidomide case lead to the first European
Community pharmaceutical directive issued in 1965, namely Directive 65/65/EEC1. No medicinal product
should ever again be marketed in the EU without prior authorisation. On 16 July 1998, the FDA announced the
approval of Thalidomide for Hansen's Disease
(Leprosy) for erythema nodosum leprosum (ENL). This imposed unprecedented authority to restrict distribution
(Thalidomide Education and Prescribing Safety oversight program- S.T.E.P.S).
Several notorious cases of adverse events have been widely publicized in recent years. One is the case of cerivastatin (Baycol, a popular cholesterol-lowering drug) from Bayer. In 2001 cerivastatin (Baycol) was removed from European and USA markets because of the risk for rhabdomyolysis (Bayer, 2001; Furberg CD, 2001; Davidson MH., 2002; Kind AH, 2002; Ravnan SL, 2002; Staffa JA, 2002; Maggini M, 2004). In 2001 when the drug was recalled, there were approximately 700,000 users of the drug. The initial cost of the recall was US $20 million in refunds for active prescriptions. (Eakin, 2003) An additional US $705 million in lost operating earnings and more than US $150 million in out-of-court settlements magnified the negative financial impact.
Prepulsid was withdrawn form the market due to cardiovascular adverse effects
(Griffin JP., 2000; Wilkinson JJ, 2004).
In late 2003 there was the SSRI case, concerning the antidepressant medicines known as selective
serotonin reuptake inhibitors (SSRI). The SSRIs were associated with an increased risk of suicidal
behavior (Fergusson D, 2004; Gunnell D, 2005). In 2004 the COX-2 inhibitor
rofecoxib
(Vioxx) was withdrawn because of cardiovascular adverse effects (Dyer C., 2004; Juni P, 2004).
The sales and marketing process
The sales and marketing of drugs is also highly regulated. The Federal Food, Drug, and Cosmetic Act (the act) requires that all drug advertisements contain (among other things) information in brief summary relating to side effects, contraindications, and effectiveness. In the US, the FDA Office of Medical Policy, Division of Drug Marketing, Advertising, and Communications (DDMAC) takes care of this. One of the most important reasons for horizontal mergers in the pharmaceutical industry is to reduce the operational costs of Sales and Marketing.
Improving the process
"...If biomedical science is to deliver on its promise, scientific creativity and
effort must also be focused on improving the medical product development process itself,
with the explicit goal of robust development pathways that are efficient and predictable
and result in products that are safe, effective, and available to patients. We must
modernize the critical development path that leads from scientific discovery to the patient..."
Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products, FDA ( March 2004)
In 2000, EUFEPS established the New Safe Medicines Faster Project, the ultimate goal of which would be to contribute to effective development of medicines for the benefit of the European citizens. In a Workshop, held on March 15-16, 2000, in Brussels, ideas and suggestions for research topics, methodologies, techniques and other means of promoting the drug development process were identified, put together and published in the Workshop I Report. In the future, it was sugested, identifying new technologies, capable of more effective selection, development and approval of new, innovative and safe drugs; using such technologies to increase the capacity and speed of the pharmaceutical development process; and cultivating a pan-European interdisciplinary network to bridge the existing gap between industry, academia, health care and regulatory authorities; would to be of paramount importance.
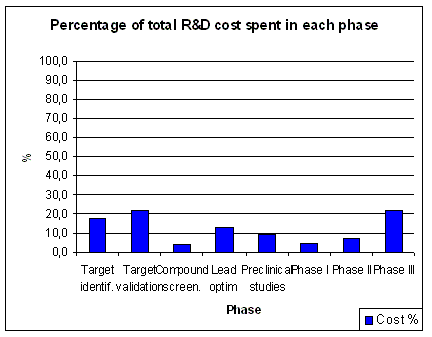
Figure 24. Budget spending as a percentage of total R&D budget (US$ 935M) Source: Life Science Insights, Ernst & Young, Tufts CSDD and Boston Consulting Group, July 2004. |
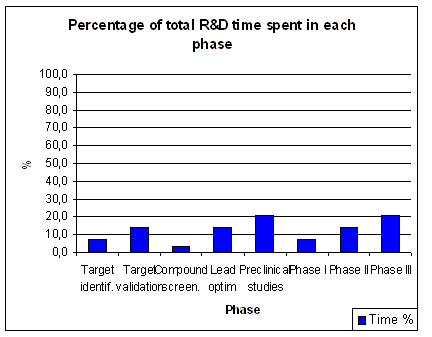
Figure 25. Time spending as a percentage of total R&D time (14.5 years) Source: Life Science Insights, Ernst & Young, Tufts CSDD and Boston Consulting Group, July 2004. |
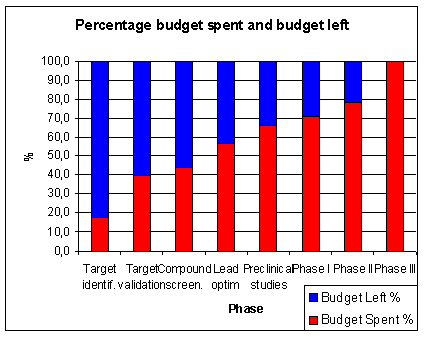
Figure 26. Budget spent and remaining as a percentage of total R&D budget (US$ 935M) Source: Life Science Insights, Ernst & Young, Tufts CSDD and Boston Consulting Group, July 2004. |
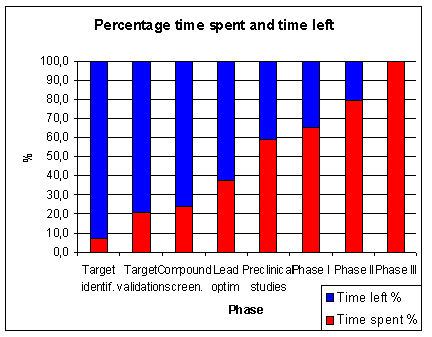
Figure 27. Time spent and remaining as a percentage of total R&D time (14.5 years) Source: Life Science Insights, Ernst & Young, Tufts CSDD and Boston Consulting Group, July 2004. |
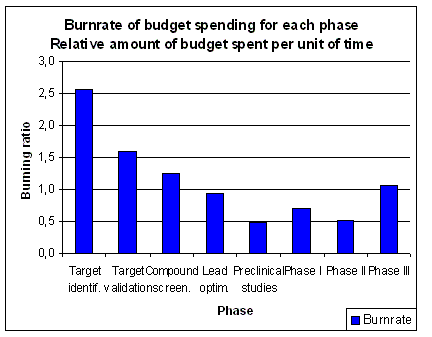
Figure 28. Burnrate of budget for each individual phase. Source: Life Science Insights, Ernst & Young, Tufts CSDD and Boston Consulting Group, July 2004. |
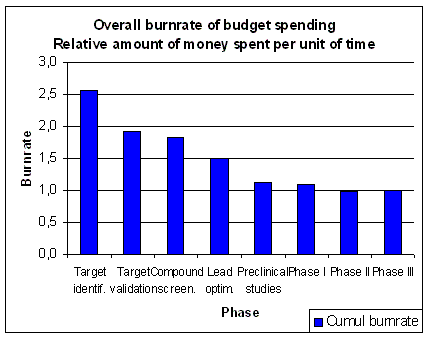
Figure 29. Cumulative burnrate of overall process. Additional impact of a phase on overall burnrate. Source: Life Science Insights, Ernst & Young, Tufts CSDD and Boston Consulting Group, July 2004. |
Every project or process has a time, cost and quality, which are important parameters when we want to improve its performance.
When we look at drug discovery and development, we look at a process which is applied on particular R&D projects.
Do we apply the right process on our individual projects?
Let us now take a look at the cost and time of the overall R&D process, which nowadays starts with target identification
and target validation. We already know that the output of the overall process is low (90% attrition in clinical development).
When we take a look at our budget (US$ 935M), we spend about 18% on target identification, qualification
and prioritization, 22% on target validation and we spend about 22% on Phase III clinical trials (Figure 24).
When we take a look at the most time consuming phases, we spend almost 21% of our time on preclinical studies and
about 21% on Phase III clinical trials (Figure 25).
By the time we are finished with lead identification and optimization, we have spent about 40% of the
R&D budget. By the time we reach Phase III of clinical development, we have spent
about 80% of our budget. From target identification to preclinical studies it takes about 66%
of our total R&D budget, which leaves us with 33% for clinical development (Figure 26).
When we take a look at the time, we spend about 60% of our time from target identification to preclinical studies, which leaves us with
40% of our time for clinical development (Figure 27).
Let us now take a look at the burnrate of our budget per unit of time. At its start the process resembles a fighter jet taking of
full throttle forward, afterburners glowing and racing towards the sky. When we reach preclinical development
the process resembles a caravan of mice and men crossing the (pre-)clinical-desert until we reach Phase III
(Figure 28 and Figure 29).
Compared to the "primitive" physiology based (empirical) process we have added a target identification and validation
step in-front of the process, which consumes about 40% of our budget and 20% of our time, but we have neglected to balance
this investment with the quality of its predictive power in relation to the clinical outcome of the process (75% failure due
to biological reasons). Improving a process requires a balance between cost, time and quality. Target identification and validation
should be done with an in-depth patho-physiological understanding of the biological process at a molecular level and not only
the target on itself. Try to understand the system of biology and the biology of the system, not only the mechanics of target-drug interaction.
In order to improve the drug discovery and development process, where should we try to optimize it? We have to balance time, cost
and quality. Adding more steps in front of the process as with target identification and validation is not an issue anymore.
Instead we should do things different and improve the time, cost and quality of what we are doing in a balanced way.
A critical path defines the optimal sequencing and timing of interventions by all stakeholders involved in a procedure
(Coffey RJ, 1992; Kost GJ., 1983; Kost GJ., 1986). Critical paths have to be developed through collaborative efforts of basic
and applied scientists, managers and others to improve the quality and value of drug discovery and development.
Unbalanced changes in a project process (scope, time, cost, quality), lead to a disproportionate decline in performance.
Quality should be measured against the impact on clinical success and not only on the next step in the process. After about
7 or more years in pre-clinical research, a new drug is ready for filing an initial new drug
application (IND) after which the FDA's Center for Drug Evaluation and Research (CDER)
monitors the clinical studies. The CDER monitors the study design and conduct of clinical
trials to ensure that people in the trials are not exposed to unnecessary risks.
The Center for Biologics Evaluation and Research (CBER) is the Center
within FDA that regulates biological products for human use under applicable federal laws. Biologics,
in contrast to drugs that are chemically synthesized, are derived from living sources
(such as humans, animals, and micro-organisms). The FDA monitors the participants of clinical trials
(FDA/ORA Bioresearch Monitoring Information Page).
In Europe the European Medicines Agency (EMEA) is a decentralised
body of the European Union with headquarters in London. The
Committee for Medicinal Products for Human Use (CHMP),
deals with medicinal products for human use. In Europe the EMEA is the bridge between the pharmaceutical industry
and the national "Competent Authorities". In Europe an Investigational Medicinal Product (IMP)
is the name for a drug in clinical development. In Europe a Development Medicinal Product (DMP) is a medicinal
product under investigation in a clinical trial in the EEA, which does not have marketing authorization in the
European Economic Area (EEA).
The clinical trials, from phase I to III are highly regulated and a company can only optimize the flow of events, but up to a large part it cannot decide freely what needs to be done in these stages of the process (e.g. ICH E6 Good Clinical Practices). The ICH develops guidances for harmonisation of drug development on Quality (Q), Safety (S), Efficacy (E) and Multidisciplinary (M) topics. Once a drug hits a regulatory authority, such as the FDA (CDER) or the EMEA strict rules need to be followed for the approval and failure to comply will only delay this process. The European legislation on pharmaceuticals can be found in EudraLex - The Rules Governing Medicinal Products in the European Union
So it is by improving the quality and shortening the process in drug discovery an preclinical development, a pharmaceutical company can make the most significant difference. But this has proven to be a dauting challenge up to now, as attrition rates in clinical trials remain high. A reduction of more than 60 % in time and about 50 % of the costs could be achieved by implementing a well-designed e-Clinical process (people, process, technology and proper change management), buth this does not yet deal with the fact that about 9 out of 10 INDs (USA) or IMPs (EU) do not belong in clinical development at all.
A lot of money is being lost in drug development and clinical trials because there are too many drugs in clinical trials which should have never reached this stage. Every approved NDA carries the burden of all the other INDs which failed and with 9 out of 10 INDs faling, this burden is very high. This shows that the gatekeepers of (pre-)clinical drug development are failing, which should not happen (in such high numbers) in a well-established stage-gate process. The stages provide the information for the gatekeepers to decide, but when the predictive power of stage-based data is too low, the decisions at the gates are of limited power. The results in drug discovery and preclinical development are biased towards overestimating the chances of success in clinical development. Efficacy is overestimated and adverse effects are underestimated. There is a need for a broader strategy to support go-no go decisions at each stage-gate. The failure to stop 90% of candidate drugs before IND filing, only becomes visible years later in drug development. Late stage attrition in drug development is due to early stage failure of disease models in drug discovery and preclinical development.
Improving drug development
Evolution of the overall process
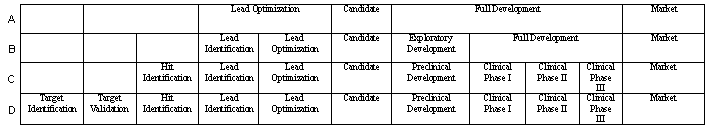
Figure 30. Evolution of discovery and development process A. 1950s and 1960s, B. 1980s, C. and D. 199Os and present. Modified from Ratti E., 2001. |
The drug discovery and development process has changed considerably over the past 50 years (Figure 30).
The discovery process had several steps added in-front which were meant to reduce uncertainty and make the overall process
more predictable. Clinical development was divided in multiple stages, but the true proof of therapeutic improvement
for a given therapy compared to either placebo or competing therapies is still at the end of the pipeline, now in Phase III.
The discovery and preclinical development stages cannot answer the questions of clinical development.
What happened up-front is that we moved further away from man and moved down to the single molecular level. We still cannot
model the complexity of man, but we can model a molecule. We reduced complexity and increasingly introduced false positives
and poor data quality. The latest developments are to bring man, the ultimat model organism, back into the process
in an earlier stage (e.g. Phase 0, microdosing). Much work remains to be done to improve the predictive power of those early stages.
The early stage predictions of success and failure in relation to late stage development should capture more of the
complexity of pathological processes in man into the models employed. What happens can be compared to what is going on
in the poem "The Blind Men and the Elephant" by John Godfrey Saxe.
A lot of detail, but no understanding of the complexity of the overall behavior of the drug in relation to its place in
the "ecological" system of the "biotope" man. The focus on molecular targets in recent
years, now resembles the situation in the poem "Der Zauberlehrling"
from Johann Wolfgang Goethe. The molecular "Sorcerer's Apprentice" can no longer control the spirits that
he called and now needs help to master the deluge of new and unvalidated targets.
We added inner resolution (molecular instead of system level), but at the same time we reduced the outer resolution
(molecular resolution instead of system-wide overview). Man is not a pile of molecules, but a complex ecosystem.
Process performance
The process does not perform at the same historical success rates anymore as attrition has now reached 92%
Preclinical and clinical development is a process driven endeavour where the improvements can be made
by improving the process management, both in management approach as well as with
better project management tools. Model improvement in preclinical development is a crucial issue.
The main reason for failure in clinical development is due to the failure of preclinical models.
The current bottlenecks in drug development are:
- Predictive pharmacology (PK/PD).
- Predictive toxicology (Tox).
- Lack of validated biomarkers.
- New clinical trial designs.
Pharmacokinetics (PK) describes the kinetics of a drug, or how the body
handles a specific compound. Generally, it involves the absorption of the compound, where the
compound goes in the body, how the compound is changed, and how it is eliminated:
absorption, distribution, metabolism, excretion (ADME) (Bohets H, 2001; Caldwell G.W., 2004; Parrott N, 2005)
Pharmacodynamics (PD) or drug metabolism (DM) describes the impact that the drug has on the body, i.e. what are the drugs
effects on the body? Pharmacodynamics (PD) studies
the relationship of the time course of a drug (and metabolites) in the body and its effects,
it describes the action of a specific compound with regard to its uptake, movement, binding and
interactions at its site of activity. A general way to consider these is pharmacokinetics (PK) is
what the body does to the drug, and pharmacodynamics (PD) is what the drug does to the body.
Reactions involved in drug metabolism (DM) are often classified as Phase I (activation) and Phase II (detoxification) reactions. Enzymes catalyzing Phase I reactions include cytochrome P450 enzymes. Enzymes catalyzing Phase II reactions include the conjugation enzymes UDP-glucuronosyltransferases (UGT), glutathione S-transferases (GST) as well as other enzymes that protect the cell from toxic damage due to oxidative stress. Phase I and Phase II enzymes acting in concert, convert hydrophobic compounds to more hydrophilic compounds that can be readily eliminated in bile or urine.
Preclinical development
Once a chemical lead is discovered, it is subjected to preclinical testing to assess biological activity.
Preclinical studies are conducted both in vitro- in cell cultures and tissues- and in vivo- on live
animals such as dogs, monkeys, and pigs. In addition to establishing the drug's pharmacological effects,
these studies also identify acute and subchronic toxicology, teratogenicity, and carcinogenicity risks.
How to find out if a discovery lead has the physical and chemical, as well as the biological,
properties to be a valid drug development candidate? Many disciplines are involved in hit-to-lead
transition and lead development. From determining Quantitative Structure Activity Relations (QSAR) to in-vivo
assays in model organisms. The process of lead optimization is an iterative process where many
scientific disciplines are involved of which I only mention a few. The problems with late stage
attrition in clinical development has its cause in the decisions made at the transition
from "model to man". We are unable to predict clinical success from preclinical disease
models in 90% of all drugs in clinical development.
Six scientific disciplines are involved in
preclinical compound characterization:
- Analytical and bioanalytical methods
- Pharmacology (e.g. therapeutic ratio, Mode Of Action)
- Nonclinical formulation
- Pharmacokinetics (PK, ADME)
- Pharmacodynamics (PD, DM)
- Pathology and toxicology (Path/Tox)
This is the traditional matrix of techniques involved in preclinical assessment of a drug candidate (pharmacokinetics (PK) and pharmacodynamics (PD) are of course also being studied in patients during clinical development). The final decisions concerning the usefulness of a drug are the domain of experimental and clinical pharmacology (Burger A., 1987). Bioavaiability of a drug is an important issue, as so elegantly captured in Lipinski's rule of five and can be used as a rule of thumb to indicate whether a molecule is likely to be orally bioavailable (bioactive) (Lipinsky CA, 1997). However this has not been able to reduce the late stage attrition rate in clinical development. Most of the tools used for toxicology and human safety testing are decades old and may fail to predict the specific safety problem that ultimately halts development or that requires post authorization withdrawal. Each aspect of preclinical safety studies (pharmacological screening for unintended effects; pharmacokinetic investigations in species used for toxicology testing; single- and repeat-dose toxicity testing; and special toxicology testing (such as mutagenicity) has not been rigorously tested by a robust analysis of its predictive power. Preclinical development was never designed to make up for the pathophysiological deficit of target-based drug discovery. Physiologically unvalidated development candidates were mainly screened for pharmacokinetic (PK) properties and pharmacodynamic (PD) properties, but this does not validate their clinical therapeutic efficacy in a clinical patho-physiological environment.
One reason for this stagnation of
inovation in preclinical development is the fact that many of these experiments are required and highly regulated by
regulatory authorities on IND and NDA filing (Hayashi M, 1994; Legler UF., 1993; Spielmann H, 2001). The
Organisation for Economic Co-operation and Development (OECD) has provided
many guidelines, such as the Reproduction/Developmental Toxicity Screening Test (OECD Guideline 421).
With limited resources, a company must focus on
those tests which it has to perform to get the clinical development candidate accepted in
the first place. There is a trend to improve the preclinical evaluation of drugs, such as performing Phase 0 tests.
Regulatory authorities are also aware of the fact that something has to happen to reduce attrition rates
in clinical development. The focus is on optimising the interface between late preclinical development and early
clinical drug development by utilising modern in vitro - in vivo extrapolation techniques. The industry has to
improve the current wasteful and uninformative system
for testing drug candidates, and shift to research methods that use biomarkers to predict
drug side effects and benefits (derived from a speech given by FDA acting deputy director Janet Woodcock).
Biomarkers could help us to to improve the predictive power of drug discovery and early drug development
(Fowler BA., 2005; Kola I, 2005). Verification that a biomarker assay is specific for its intended purpose
poses a formidable challenge.
We need (validated) biomarkers for preclinical and clinical development in order to:
- Treat diseases more effectively:
We currently lack predictive biomarkers to stratify patients with similar diseases as well as accurately measure disease susceptibility, presence and progression. - Verify the impact of novel drugs on targets/pathways:
We currently lack the ability to determine the ability of a novel drug to bind the desired target and whether this binding actually leads to changes in the desired pathway. - Avoid Serious Adverse Events (SAE):
About 1.5 million people are hospitalized each year due to adverse effects of prescription drugs. - Increase drug development predictability:
Only 1 in 10 drugs entering Phase I ever reach the market with the great majority of compounds failing in Phase II.
We really need drug candidates (NCEs, NBEs) which make it to the market at much higher rates than with the current 10% overall success rate from IND to NDA. This will require a significant paradigm change in the assessment of potential investigational drug candidates (Apic G., 2005; Caldwell GW, 2001; Schadt EE, 2005; Shaffer C., 2005).
Highly sensitive techniques, such as Accelerator Mass Spectrometry (AMS) and PET allow for the detection of biomarkers. Accelerator mass spectrometry (AMS) is a mass spectrometric method for quantifying rare isotopes, which is being applied to biomedical and toxicological research (Barker J, 1999; Brown K, 2006; MacGregor JT, 1995; Turteltaub KW, 1990). AMS can be used to study long-term pharmacokinetics and to identify biomolecular interactions in neurotoxicology and neuroscience (Palmblad M, 2005). AMS enables compounds and metabolites to be measured in human urine and plasma after administration of low pharmacologically or toxicologically relevant doses of labelled chemicals and drugs (White IN, 2004).
Clinical development
For many scientists working in drug discovery and preclinical development, the environment in which
clinical trials happen is still something mysterious. I want to clarify some of these issues,
mainly the regulatory (and scientific) framework in which these complex and expensive trials have to be performed.
Drug discovery and preclinical development should be done while keeping an eye on clinical development.
Maybe this helps to understand the fears and nightmares of those scientists and their collaborators,
every time they start a development track and face the fact that up to 90% of their efforts are in vain.
A clinical trial (also clinical research)
is a research study in human volunteers to answer specific health questions. Carefully conducted clinical
trials are the fastest and safest way to find treatments that work in people and ways to improve health.
Interventional trials determine whether experimental treatments or new ways of using known therapies
are safe and effective under controlled environments. Observational trials address health issues in large
groups of people or populations in natural settings. Each clinical trial starts with the definition of
a primary question (study hypothesis). From that hypothesis the best way to confirm or reject it,
can be designed. There are several designs possible, which can be found in the literature on clinical trials.
A randomised, double-blind, crossover or factorial, multi-site Phase III
clinical trial is a complex endeavour and a drug which fails at this stage has cost an enormous
amount of money (Figure 31). Not to mention the shattered hopes of the patients it was intended to provide with new hope.
A Phase III clinical trial may involve up to 5,000 patients distributed over numerous clinical trial sites,
which gives you an idea of the logistic complexity to manage such a trial. It is important to keep in mind
that a Phase I trial does not deal with the outcome of a Phase II trial and so on. Each trial type is
designed to answer a different type of question and it only provides
an answer to this question(s), not to the ones being dealt with in another type of clinical trial.
Clinical trials are becoming more expensive and even more regulated. A Phase I trial costs about
US$ 8,000-15,000/subject, a Phase II costs about US$8,000-15,000/patient and finally
a Phase III trial costs about US$4,000-7,500/patient. Improving a proces is a matter of methodology and technology.
The overall profile of therapeutic indications and adverse events, leads to the labeling of the proposed drug. The "sponsor" of a new drug must obtain approval from the FDA by specifying both the medical conditions the drug is effective against and the patients groups for whom the drug has been shown to be effective. This information is contained in the proposed "label" submitted by the developer or sponsor. It is the sponsor's responsibility to assemble all the evidence that would support the uses proposed in the label (preclinical and clinical development). With the wide gap between molecular targets and clinical diseases, this has become increasingly complex and risky. This also explains the problems with "first-in-class" drugs and the reduced risk once knowledge and understanding build after years of widespread use. There is also the potential safety problem of "off-label" use of a drug, besides the problems with reimbursement.

Figure 31. Clinical trials from Phase I to Phase IV. Phase II can consist of a Phase IIa and IIb. Phase III can also consist of a Phase IIIa and IIIb. |
A basic clinical trial process consists of several stages:
- Hypothesis formulation, primary and secondary endpoints
- Protocol development
- Investigator/site selection and trial preparation
- Subject identification and enrollment (causes most of the delays)
- Collection, monitoring and processing of data (CRF, PRO, Lab)
- Clinical trial management
- Data analysis and reporting of results (SAS)
- Submission for review by a regulatory agency (FDA, EMEA, ...)
A "ram-it-through paradigm" in clinical trials readily produced beta-blockers, H2 blockers, nonsedating antihistamines, and other big classes of drugs. The development of predictive models to assist the decision process to enter the next Phase of clinical development is an interesting path taken to reduce late stage attrition (Albert JM, 1994; De Ridder F., 2005; Hale M, 1996; Holford NH., 2000). There is a transition going on from empirical to causal models for deriving evidence of effectiveness. There is also a transition from empirical to causal models for deriving evidence of safety. Clinical development is changing from a reactive empirical model to a proactive Model Based Drug Development (MBDD) process (pharmacometrics). Instead of rushing through clinical development, a learn-and-confirm approach allows for a more dynamic and adaptive process (Sheiner LB., 1997). Phase I and IIa are the learning phases and Phase IIb and III are the confirming phases. There is a clear trend to learn more earlier in the drug development process. Phase I is no longer just for establishing safety and dosing levels, Phase I research is playing an increasingly important role obtaining more data about the potential success of a drug. The emphasis is increasingly on mechanistic early phase clinical trials to maximise the chances of obtaining clinical data to make sound go/nogo decisions. Model-based drug development includes exposure/response assessments in the form of pharmacokinetic/pharmacodynamic (PK/PD) modeling. Pharmacokinetic (PK) and pharmacodynamic (PD) modeling and simulation (M&S) are powerful tools that enable effective implementation of the learn-and-confirm paradigm in drug development (Chien JY, 2005; Gobburu JV, 2001; Grasela TH, 2005). One of the major prerequisites for the successful application of PK/PD-modeling, however, is the availability of response measures such as biomarkers that provide an immediately accessible link between pharmacotherapeutic intervention and clinical outcome and allow to easily assess variations in desired and/or undesired drug effects in response to changes in dose, dosage regimen, dosage formulation, administration pathway, or external factors affecting drug response. Biomarker-based PK/PD modeling can become the basis for a scientifically driven, evidence-based, streamlined drug development process.
The problems with drug discovery and development are not limited to scientific and technical issues alone.
Discovery and preclinical research may be a scientific and technical minefield, but with clinical development we in addition
enter a moral and ethical minefield. Life in the lab may not be easy, but life at the bedside isn't either.
Improving the clinical development process is not easy, as we have to operate in a highly regulated environment,
which limits the freedom to change the process. In a highly regulated environment you cannot do different things, but you
have to do (regulated) things differently, e.g. a new statistical approach (e.g. Bayesian methods), modeling and simulation,
learning and confirming (Maurer W., 2005). In the early stages of drug development we should be able to extract more
predictive information from our research, to reduce the late stage attrition in Phase II and III. More knowledge and
understanding of complex processes earlier on, would allow for better predictions and less failure later on in the process.
Applied science within regulatory constraints is the only way to bring basic science into clinical reality.
Just to give you an idea about all the regulatory issues involved, I will give an overview of some guidelines (Grunfeld GB., 1992).
As clinical trials involve human experimentation, they are to be conducted according to high ethical standards.
Historical events lead to the
adoption of ethical guidelines for the conduct of research on human subjects.
The Nuremberg Doctors' trial,
officially United States of America versus Karl Brandt, et al., lead to the Nuremberg Code.
The Nuremberg Code (1947)
laid down 10 standards to which physicians must conform when carrying out experiments on human subjects.
In summary, the Nuremberg Code includes the following guidlines for researchers:
- Informed consent is essential.
- Research should be based on prior animal work.
- The risks should be justified by the anticipated benefits.
- Research must be conducted by qualified scientists.
- Physical and mental suffering must be avoided.
- Research in which death or disabling injury is expected should not be conducted.
Government agencies take care of enacting the laws and regulations, such as the US
Food and Drug Administration (FDA)
(e.g. CBER Guidances), The European
European Medicines Agency (EMEA) and the Japanese
Ministry of Health, Labor and Welfare.
On 1 April, 2004, Japan's
Pharmaceuticals and Medical Devices Agency (PMDA)
began conducting NDA reviews for the Ministry of Health, Labor and Welfare.
The Council for International Organizations Of Medical Sciences (CIOMS) develops principles
and proposals for the collection, evaluation, and reporting of safety information obtained during clinical
trials to all appropriate stakeholders. The CIOMS I workgroup (1990) dealt with international reporting of
Adverse Drug Reactions (ADR) and created the "CIOMS I reporting form" for standardized international reporting
of individual cases of serious, unexpected adverse drug reactions.
As regulatory authorities increase their standards on drug development, they put the hurdle higher
for all the players in the field. So, all companies face the same challenge to improve their process.
One of the positive consequences has been that the US pharmaceutical industry is now the most competitive
in the world, as they had to adher to increasing quality standards. The US pharmaceutical market is still the
most profitable of all, but also one the most regulated.
As clinical trials in Phase III are large-scale projects, streamlining data exchange procedures can make a significant contribution to cost reduction and shortening the duration of a trial, e.g. for a Computer Assisted New Drug Application (CANDA) of the FDA. Standardization of data formats is one aspect of process improvement for which many attempts have been made over the years, e.g. Drug Application Methodology with Optical Storage (DAMOS), Multi Agency Electronic Regulatory Submission (MERS), Market Authorization by Network Submission and Evaluation (MANSEV) and Soumission Electronique des Dossiers d'Autorisation de Mise sur le Marché (SEDAMM). The need for international standardization in clinical trials is being dealth with by organizations such as the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), e.g. Common Technical Document (e-CTD for e-submission). There is the ICH E3 guideline on the Structure and Content of Clinical Study Reports. The Clinical Data Interchange Standards Consortium (CDISC) develops XML-based standards for data exchange in clinical trials (e.g. ODM, SDTM,...). eSignatures should allow for exchanging digital data in a secure way within the pharmaceutical industry. The Secure Access For Everyone (SAFE) initiative, is a set of standards for digitally signed transactions that create a trusted community for legally enforceable data exchange.
Clinical trials are process driven and as such amenable to process management improvement. Improving clinical
trials is an ongoing effort, driven by the tendency to new clinical trial designs and e-Clinical trials.
The traditional Randomized Controlled Trial (RCT) is still the standard, but new designs are being applied.
Besides a new design, new technology can help to improve the clinical trial process.
The Internet provides a means to improve the process for large multicenter clinical trials (Paul J, 2005).
However clinical trials from Phase I to III (and IV) are a highly regulated process,
(FDA Title 21 CFR part 11,
HIPAA,
GCP,
GMP,...) which limits the freedom for improvement
(e.g. Computer System Validation). Clinical trials gain
speed by using Cinical Trials Management Software (CTMS), Clinical Data Management Software (CDMS) and automating the
process of data capturing (RDC or Remote Data Capture and
EDC or Electronic Data Capturing), such as for the Patient-Reported Outcome (PRO) and Case Report Forms (CRF).
The usage of computer systems in clincal trials is guided by the
FDA Guidance for Industry on Computerized Systems Used in Clinical Trials.
The Biomedical Research Integrated Domain Group (BRIDG) Model is a comprehensive
domain analysis model representing biomedical/clinical research. It was developed to provide an overarching model
that could readily be comprehended by domain experts and would provide the basis for harmonization among standards
within the clinical research domain and between biomedical/clinical research and healthcare. Work is going on to
develop a Clinical Trials Object Model (CTOM) and a Structured Protocol Representation, from which e-Clinical trials could be
designed, developed and managed.
The procedures for data submisson are being improved by
Electronic Regulatory Submissions and Review.
The review process is being regulated by Good Review Practices (GRPs).
The FDA JANUS project will combine information from numerous clinical trials into a single data warehouse. JANUS will give the
FDA the ability to cross-analyze data from multiple trials, identify systemic deficiencies across similar products,
and detect potentially dangerous drug interactions. So, a lot of improvements are going on in the drug review process,
but this will be of limited success if the drugs (NCE, NBE) themselves do not show better quality profiles.
Process improvement is not only a matter of using ICT, but also requires a change in project management principles in order to succeed. Clinical trial process improvement is for 80% a matter of people and process and for 20% a matter of technology (ICT, biomarkers,...). An overall improvement from protocol (e.g. eProtocol) to NDA submission is required, for instance by shortening the time from protocol completion to First Patient First Visit (FPFV), First Patient In (FPI) to Last Patient Out (LPO), Last Patient Last Visit (LPLV) to DB lock and finally from DB lock to NDA. A reduction of more than 60 % in time and about 50 % of the costs could be achieved by implementing a well-designed e-Clinical process (people, process, technology and proper change management). An e-Clinical trail also allows for better process monitoring (performance metrics system).
Improving drug discovery and preclinical development
The modern pharmaceutical industry faces
an increasingly widening gap between, on the one hand, growing numbers of potential drug targets
and lead compounds and, on the other hand, a lack of reliable methods to identify those
molecular targets unequivocally linked to disease pathophysiology and lead compounds with
the best chance of success in (pre)clinical development. How should we proceed to
improve drug discovery and preclinical development? We have seen an enormous investment in research at the
infra-cellular level, such as High Throughput Screening (HTS), genome based and proteome based disease
models in the past ten years and at the same moment have witnessed a
disproportional decline in the productivity of research and development in drug
discovery (Horrobin DF, 2000; Horrobin DF, 2001; Noble D., 2003; Bleicher KH, 2003, Betz UA, 2005; Bilello JA., 2005). The pharmaceutical industry has yet to find a
way to reduce its high attrition rates (
The consolidation in the pharmaceutical industry will not solve this problem in the long run, as it only
reduces the costs (mainly of sales and marketing and manufacturing) but does not improve scientific productivity; it only
postpones the moment of truth. The scientists themselves will have to find new
ways to improve their productivity; management cannot do this in their place.
Society tries to protect itself against the adverse effects of new drugs, such
as with Thalidomide in the sixties (McBride WG, 1961). This is done by
increasingly stringent regulations but the currently used methods in the
discovery process for new drugs cannot keep pace with these new requirements.
However, as we can see, increasingly strict regulations do not explain all the
problems pharmaceutical research is facing today.
We can summarize the (r)evolution in the overall drug discovery and development process as such:
Traditionally we have a "trial and error" approach:
This will have to evolve into a "cognitive" chemical biology approach:
Disease models in drug discovery and preclinical development
Scientific progress from basic science to its applications
Basic and applied research achieve results through brilliant ideas and hard work.
"Adde parvum parvo magnus acervus erit" (Ovidius, By adding little to little
there will be a great heap). You cannot get much done in a short time, but
once you start to string together work for several years or decades, you have the start
of a body of work that puts a mark on the world.
The concern and goal of these articles is to clarify the problems with bringing basic
research to clinical applications, not to criticise basic research as such. When we look at
scientific methodology not from the perspective of understanding fundamental biological processes,
but for their predictive power to generate results which facilitate the application of basic
science to clinical reality, a different picture emerges. I want to look at scientific methodology
with its impact on treating the pathological process in man as a reference.
Keep in mind Occam's Razor: "Numquam ponendo
est pluritas sine necessitate", which we should translate as "One should use
the explanation that is simple enough to explain all there is to explain, but nothing simpler".
Quite often our explanations fail to capture the entire biological phenomenon, which is why we fail in almost 90%
of all drugs in clinical development. 75% Of all failures in clinical development are caused by lack of
efficacy (30%), adverse efects in man (13%), animal toxicity (20%) and pharmacokinetics (12%).
I will take a look at several aspects of the discovery and development process to provide wide ranging information
to those who are interested in improving the pharmaceutical R&D process. I want to avoid "stovepiping" by skipping information
levels. In 2003 Seymour Hersh wrote an article in the New Yorker titled,
"The Stovepipe". Hersh defines "stovepiping"
as taking a request for action arising from intelligence "directly to higher authorities without the information
on which it is based having been subjected to rigorous scrutiny." Stovepiping results in intelligence failures
when conclusions are allowed to pass rapidly from the lowest levels of the intelligence gathering apparatus,
the ones with their hands directly on new information as it comes in at ground level, up to the decision making
authorities many levels above without passing through the normal many-layered time-intensive vetting and
checking processes in-between. This can also be applied to a request for improvement
of the "war against disease".
Failing disease models in drug discovery
and preclinical development
Anyone working in science must realize that theories, models and approximations are powerful tools
for understanding and achieving research and development goals. The price of having such powerful
tools is that not all of them are perfect. This may not be an ideal situation, but it is the best that the
scientific community has to offer. Each individual must try to attain an understanding of the
nature of our descriptions of the physical world and what results can be trusted to any given degree of
accuracy (see also
Models, Approximations and Reality).
Currently used disease models fail to predict the outcome of clinical development and are incapable to
reduce attrition rates in drug development. The reasons for failure can be summarized as follows.
Drugs fail in development and beyond due to:
What should we do to improve this?
There is no single disease model which
allows us to predict clinical success in all cases. Bridging the gap between molecule
an man is a delicate process, which requires careful consideration and design. Quite often
there is not enough consideration of building and validating the path from clinic to
model and back. In the end it is clinical
reality which decides on the fate of new drugs and not the technology or
disease models used to create them. The successful identification of drug
targets requires an understanding of the high-level functional interactions
between the key components of cells, organs and systems, and how these
interactions change in disease states (Butcher EC, 2004; Noble D, 2003; Stumm G, 2002). Pre-clinical studies and early clinical
trials should pay more attention to both the pharmacology of the drug as well
as the (in-vivo) biology of the target (Newell DR., 2005). Both basic and
applied research have have made truly enormous contributions to the health
of mankind, but the challenges ahead are no less than they were in the past.
Only with an open mind and a thorough analysis of the present situation
we will be able to analyze the problems of present day research and
development.
Validating the predictive power of our disease models is not an easy task as we operate in a complex
pathophysiological environment.
"...One must rely heavily on statistics in formulating a quantitative model but, at each
critical step in constructing the model, one must set aside statistics and ask questions.
... without a qualitative perspective one is apt to generate statistical unicorns,
beasts that exist on paper but not in reality.
... it has recently become all too clear that one can correlate a set of dependent
variables using random numbers as dependent variables. Such correlations meet
the usual criteria of high significance. ..." (Hansch C, 1973).
We fail to predict clinical success with our current disease models, which translates
itself in a high (up to 90%) attrition rate in clinical drug development.
The problem of prediction in relation to a model system (van Drie JH., 2003) (free from the "Kubinyi Paradox"):
There are two approaches to drug discovery,
historically a physiology based approach was used, while nowadays a target based approach
has become more popular. Most of the problems of the drug discovery and development process can be
traced back to its early stages, namely target validation and lead selection.
With respect to the targets this is, among other factors, due to a lack of definite clues with
regard to the (dis)regulation of cellular pathways that underlie diseases. This gap in basic
knowledge is even further widened by the lack of adequate animal test models and
disease markers to monitor the particular disease process as well as the outcome of
therapeutic interventions. With respect to lead selection there is a poor insight in the
fundamental relations between the physico-chemical characteristics and the biological properties
of the drug candidates, including mechanisms of cellular action and toxicity, processes of
drug disposition (ADME) and the aspect of drug safety on a longterm basis. In modern drug discovery
the early stages of drug discovery involve the identification and early validation of a
disease-modifying target (Lindsay MA., 2003; Schneider M, 2004). Failing to
make the right decision at the important step of hit to lead transition has
costly time and resource implications in downstream drug development (Alanine
A., 2003; Kuhlmann J, 1999). Why do the early stages
of drug discovery fail so often and why are they the cause of a huge efficiency
deficit later on in the drug discovery process?
Assessing the drugability of a target is only one of the important criteria to consider.
Drugability of a target is the feasibility of a target to
be effectively modulated by a small molecule ligand that has appropriate
bio-physicochemical and absorption, distribution, metabolism and excretion
properties (ADME) to be developed into a drug candidate with appropriate properties
for the desired therapeutic use. The weak spot is the definition of
therapeutic use, which quite often does not mean we are working with a clinicaly validated target.
Since the early successes of compound screening against isolated molecular targets in the 1970s, the industry moved away from physiological based screening to a target-based screening (Luyten W, 1993; Herz JM, 1997). In the beginning Target-based screening was initially used to improve the drug-like properties and selectivity of pharmacologically active products. Target-based drug discovery has been very successful when applied to already physiologicaly validated targets of existing drug. Later on the hope was that sequencing the human genome would generate a wealth of new targets, and the hope of almost directly linking 'genes-to-drugs' was embraced by the industry in the 1990s. When the drug discovery process moved beyond historical targets, however, it became apparent that the target-directed approach was flawed: without solid biological validation, target-based drug discovery has proven very disappointing (Williams M., 2003; Butcher E., 2005). Improving target identification requires a dramatic improvement of our understanding of cellular pathways underlying pathogenesis and/or pathophysiology. Improving lead selection requires an hypothesis-driven approach in which, out of the multitude of potential targets, a rigid selection of druggable targets is made.
There is a fundamental
problem with studying disease-relevant mechanisms in the current disease models
as the pharmaceutical industry has been investing heavily in studying the
'bricks', instead of looking at the 'building' (cytome, organism) as a dynamic
unified pathophysiological system.
Finding a gene or a target
does not equal understanding a clinical disease process in man. The genome has yielded a series
of novel molecules that do not have 20 or 40 years of biology behind them for us to understand
exactly what they do and where to apply them. The emphasis in recent
years has been on increasing quantity while at the same moment sacrificing the
quality of correlation with clinical reality. Accepting the perceived truth
that ubiquity equals utility, organisations often initiate efforts to automate
processes, assuming that improvement will be a natural consequence of automation. This avalanche of data
does not lead to an improvement of understanding.
"Le savant doit ordonner; on fait la Science avec des
faits comme une maison avec des pierres; mais une accumulation de faits n'est pas plus
une science qu'un tas de pierres n'est une maison." Henri Poincaré (1854-1912).
We increased our capacity in genomics and proteomics, but we did not improve the quality of the preclinical
physiological disease process evaluation in the same way. Technologies such as genomics
and proteomics have produced an explosion of new poorly validated targets that
actually increase the rate of compound attrition and the costs of R&D.
You could also think of it as a
pointillist painting, of which we have been looking at the individual dots,
instead of looking at the entire painting. Another analogy is that we are
trying to explain the tidal patterns of the oceans, by studying a water
molecule and ignoring the moon. We have to look for the picture that is in the puzzle early on,
not just giving the 'pieces' to the next process down the line.
We have to look at biological phenomena at several
scales of integration and from a functional point of view in order
to get a grip on the development of pathological processes. Crosslinking and
crossreferencing biological organisational levels in order to understand the
'web' of biological interactions in a pathophysiological process.
We should try to
understand the dynamics of disease processes also at a higher level of biological
integration, closer to the clinical reality, than only the genome or proteome. An
integrated cellular and organism-level approach is needed to study disease
processes (Lewis W., 2003).
When we modify a gene, e.g.
by creating transgenic animals, we must try to understand the dynamics of the
pathways we are modifying. The gene products are part of a delicate web of
intertwined pathways where subtle changes can have unpredictable effects.
Quite often transgenic animals or animals with gene
knock-outs do not show the expected phenotype, because of a different genetic
background and the highly dynamic interplay of metabolic pathways and
environmental influences on the final phenotype (Sanford LP, 2001; Pearson H.
2002).
The (early stage) disease
models we use don't work as they should do and do not provide enough overall predictive
power in relation to clinical reality. One can study cellular components, like DNA and protein as such, but
this will not reveal the complex interactions going on at the cellular level of
biological integration or in other words, the cytome.
Both medicine and pharmaceutical research would benefit from using more (primary)
cell oriented disease models (in-vivo and in-vitro) and even higher-order models, instead of using
infra-cellular models to try to describe complex pathological processes at a
molecular level and getting lost in the maze of molecules which are the building
blocks of cells. Keep in mind that cytome-oriented research is not the same as
cell-based research.
An important moment in the
drug discovery and development pipeline is the transition from discovery
research to clinical development. Different approaches to develop
gatekeepers have been proposed to reduce the failure rate in drug development
on both sides of the transition (Lappin G., 2003; Nicholson J.K., 2002;
Pritchard J.F., 2003). Translational medicine is emerging as a gatekeeper
for evaluating drugs when they traverse the "great divide" between "bench and bedside"
(Fitzgerald GA., 2005). Translational Medicine can be defined as an interactive process
between preclinical and clinical investigation. Translational biomarkers and molecular profiling
should assist in increasing success in clinical development. However, translational medicine alone
will only bring bad news earlier in the process, so it should be combined with a
concomitant improvement of disease models in drug discovery and preclinical development.
Drug discovery and preclinical development should improve the quality of drugs they allow
to enter clinical development and clinical development should be able to protect itself
from drugs likely to fail in phases I to III.
A better quality of drugs entering drug development is needed,
not just more quantity (drastic reduction of false positives). Failing in larger numbers
will not bring the solution to create a better process from discovery to phase III an IV.
If we just drive more drugs into clinical development, but keep failing at a rate of
up to 90%, we are not helping ourselves.
A highly defined
oligo-parametric infra-cellular disease model used in High Throughput Screening
(HTS) which in its setup ignores the
complexity of higher order cellular phenomena, may produce beautiful results
in the laboratory, but fails to generate results of sufficient predictive power
to avoid considerable financial losses later on in the drug discovery pipeline
(Bleicher KH, 2003). A living (primary) cell may be a less well defined
experimental environment for the biochemist, but it will provide us with the
additional modulating influences on our disease models which are lost in
lower-order disease models. The cytome can be analyzed either in-vitro in
well designed cell-based assays or in-vivo by using for instance molecular imaging.
Metabolic variation in disease models
Preclinical models should help to identify factors that are important
determinants of intersubject variability in man. We need to make a clear distinction between our inability to elucidate
the overall molecular mechanism of a disease process and population variability in metabolic activity.
Genetic variability is only one
part of the picture, as there are multiple determinants of intra- and interindividual variation:
Real life variability should be
incorporated in preclinical disease models and not excluded. Nowadays the first stages
of drug discovery and development use genetically homogeneous disease models, which as a result
do not show the same metabolic heterogeneity of patient populations. In-vivo
variation is not an artefact of life, but a fact of life. Variability in drug response
is a complex and multifactorial phenomenon, of which genetics is only a part.
Genetic and metabolic heterogeneity is now seen as reason to exclude potential
patients from treatment, not as a consequence of the failure of drug
development. We need to make a clear distinction between our inability to elucidate
the true molecular mechanism of a disease process and population variability.
Molecular diversity in a clinical disease process leads to treatment failure because of the
wrong action is taken to modify a disease process which we do not understand.
Metabolic variation in drug metabolism is another reason for treatment failure, but
is more easy to deal with as these problems involve shared properties of drugs.
If we cannot develop drugs which will work in a genetically and
metabolically heterogeneous environment, we try to reduce the patient
population until it fits our abilities. However this micro-management of
patient populations leads to a level of complexity in disease treatments the
pharmaceutical industry, physicians and society cannot deal with in the end.
Also, excluding people from treatment, because we are unable to develop drugs which
benefit a large population of people poses ethical problems. Dose optimisation however
is an interesting path to minimize side-effects. At present there are still too many roadblocks
to achieve the goal of a better and finely tuned disease treatment. Personalized medicine will remain a distant dream,
if we do not succeed in achieving a much better understanding of molecular pathophysiology and
at the same time dramaticaly improve the drug discovery and development process.
Pharmacogenomics is being used to explain differences in drug metabolism during drug development, such as with
Cytochrome P450 (Dracopoli NC., 2003; Halapi E, 2004; Kalow W., 2004). Toxicogenomics
and genotyping are used as a tool to identify safer drugs, worthwhile to enter clinical development
(Guzey C, 2004; Koch WH., 2004; Yang Y, 2004).
There are many causes for variability
in drug metabolism. this variability can lead to an Induced Error Of Metabolism (IEOM), just as
an
Inborn Error Of Metabolism
caused by a genetic defect. Variability of a metabolic change due to a drug can have many causes:
ΔMetabolic change = ΔUptake + ΔDisease state + ΔHost state + ΔElimination
In the case of a pathogen, the variation in virulence is a cause of variation,
as well as the host defense system status (e.g. the Varicella Zoster virus which
causes Varicella or chicken pox, and herpes zoster or shingles). Metabolic variation due to
variability in drug uptake and elimination can be a serious cause of trouble.
The Cytochrome P450 enzymes are an important cause of metabolic variation in the
metabolism of drugs (Slaughter RL, 1995). Many drug interactions are a result of inhibition or induction of cytochrome
P450 enzymes (CYP450). The CYP3A subfamily is involved in many clinically significant drug
interactions, including those involving nonsedating antihistamines and cisapride, that may
result in cardiac dysrhythmias. CYP3A4 and CYP1A2 enzymes are involved in drug interactions
involving theophylline. CYP2D6 is involved in the metabolism of many psychotherapeutic agents.
Variability in drug metabolism can also be caused by food. Grapefruit juice affects
the pharmacokinetics of various kinds of drugs, the major mechanism being considered to
be inactivation of intestinal cytochrome P450 3A4, a so-called mechanism-based
inhibition (Bailey DG, 1991; Fuhr U., 1998; Saito M, 2005).
The EMEA road map and the
FDA Critical Path identify pharmacogenomics (PGx) as the emerging technology that
will enable efficient and successful drug development.
Pharmacogenomics is not yet
used to design or use early stage disease models with sufficient genetic
heterogeneity to select drug molecules which will hold their activity in a
metabolic heterogeneous environment. Genetic heterogeneity, epigenetic
modulation and metabolic variation are not taken into account in the first
stages of the drug discovery process. Optimizing a drug molecule for binding to
one particular genetic variant, imminently leads to failure in a genetically
heterogeneous patient population. Randomization in experimental design to
counteract a systematic bias in ones results involves more than sample unit
randomization patterns.
Biological variation in
heterogeneous cell or animal populations may be an unpleasant fact of life, but
it correlates better to the real conditions of the genetically and
metabolically heterogeneous patient populations. Ignoring biological variation
in drug discovery will cause failure in drug development. Using pharmaco-genomics only to exclude slow metabolizers, etc.,
from clinical trials and thereby homogenizing the trial population can lead to
a dramatic reduction in potential patient population and a decline in profit
generation potential. Finding sites to manipulate metabolism which are less sensitive
to genetic variation would improve our overall success rates.
The important phase of a drug life cycle starts when it
hits the market and we better take care that it will spend its full life cycle
to generate enough revenue to fuel the company.
Hypo- or Subcellular disease models
When molecular biology entered drug discovery in the
1980s and 1990s the dominant view on the relation between genotype and phenotype was derived from the
simple dynamics of prokaryote genetics. The preeminent French scientist and 1965 Nobel laureate
Jacques Monod, said in 1972 "Tout ce qui est vrai pour le Colibacille est vrai pour l'éléphant"
("What is true for
Escherichia coli
is also true of the
elephant"). However, the outcome of the
Human Genome Project has revealed that the processing of our genetic information is
much more complex than in Prokaryotes.
We have seen an increase in
capacity of DNA and RNA expression techniques, but their information still
delivers data up to the level of the expressed protein, but not beyond. The
quantitative chain of functional causation stops at the protein level. Higher
order spatial and temporal dimensions of cellular dynamics are beyond the reach
of these techniques. Gene expression studies do not tell you about the
functional outcome of protein dynamics and enzymatic activity in the different
cellular compartments. Up and down-regulation of gene expression, does not
inform you about the functional interrelation of the encoded proteins and their
spatial and temporal dynamics in the cell. Molecular pathways do not exist as
parallelized unrelated up-and down regulating patterns, but are highly dynamic
and intertwined modular networks (
Gleevec or imatinib or STI571 (signal transduction inhibitor number 571) is a good example
of a drug aiming at a molecular target for which the entire chain of models, from molecule
to man was thoroughly explored. The disease mechanism was well understood and the molecular
biological mechanism was well-embedded in a pathophysiological understanding (Jones RL, 2005; Mauro MJ, 2001).
This endeavor required the integration of a number of disciplines, including structural
biology, computational chemistry, structurally directed medicinal chemistry,
array screening assays, and molecular and cellular biology (Druker BJ, 2000).
Publications on the association of the Philadelphia chromosome and leukemia can be traced back to the early sixities of the
twentieth century (Benson ES, 1961; Tjio JH, 1966). In 1982 it was found that in chronic myelogenous leukemia (CML),
c-abl sequences are translocated from chromosome 9 to chromosome 22q- (de Klein A, 1982). During the eighties of the
twentieth century the molecular mechanism of the gene product, a tyrosine kinase was investigated
(Pendergast AM, 1987; Maxwell SA, 1987; Lugo TG, 1990). More than
15 years of work by scientists from all over the world was needed to understand the molecular
mechanism of the disease (this costs far more than 800M US $). Once the target was identified
and the disease process understood, the selection for drug candidates could start (Druker BJ, 1996).
Finding the gene is not enough, the hard work is to
find out about the molecular mechanism of the disease and this has not changed even now the
Human Genome Project has been completed. The work on the c-abl tyrosine kinase predates the Human Genome
Project with more than 20 years (the same goes for Herceptin). With the current state of technology
and science we may expect to see the results from the new targets coming out of the
Human Genome Project in 10 to 20 years.
Let us now look at some methods used in molecular biology, not for their value in basic research,
but for their predictive value for preclinical development (a bit unfair, I know). We must
be aware that studying a basic molecular mechanism is still far away from understanding the
clinical disease process as such. Southern, Northern and
Western blots may show the quantitative sequence of gene expression up to
protein concentration (Alwine JC, 1977; Alwine JC, 1979; Howe JG, 1981; Hinshelwood
MM, 1993). DNA microarrays give a quantitative
indication of gene expression (Barbieri RL, 1994; Schena M, 1995; DeRisi J, 1996; Jeong JG, 2004; Kawasaki ES., 2004). However finding a
positive correlation between the pattern of gene expression and a given disease
state is not the same as finding a causative relationship between (a) gene(s)
and the causation matrix of a disease (Miklos GL,
2004). Moving up to the level of the dynamics of protein expression already
demands a higher degree of sophistication in both assay design and data
analysis (Kumble KD., 2003). However, without a
functional assay on in-vivo dynamics of protein function and studying its
spatial and temporal expression patterns (process flux) in the cell
(compartments) and tissue, the functional impact on the cell remains unclear (Kriete A, 2003; Young MB, 2003; Egner, A., 2004).
Studying subcomponents of cellular
pathways ignores the functional unity of the biological processes in the cell
and the functional interactions between pathways. Studying an isolated drug
target ignores important off-target interactions which become a cause of
failure too late in drug development. Studying proteins in isolation and
uncoupled from the intracellular molecular oscillating clocks, ignores the
importance of temporal patterns (Okamura H., 2004). Without a better
understanding of the phenotypic and functional outcome in the cell, the failure
rate of the drug discovery process will remain high and very costly. There is a
predictive deficit in the current oligo-parametric disease models used in
pharmaceutical research which necessitates complex and expensive studies later
on in the drug development pipeline to make up for the predictive deficit.
A simple homogeneous
binding assay, will fail to capture important aspects of functional protein
heterogeneity. G-protein-coupled receptors (GPCRs)
represent by far the largest class of targets for modern drugs, but we have not
yet unraveled the subtle dynamics of their function
(George SR, 2002; Kenakin T., 2002; Kimple RJ, 2002; Ellis C., 2004; Kristiansen K., 2004). Heterodimerization enhances the complexity of ligand
recognition and diversity of signaling responses of heterotrimeric guanine nucleotide-binding protein-coupled
receptors (GPCRs) (Foord
SM., 2003; Liebmann C., 2004). Heterogeneity of
protein interactions that underlie both cell-surface receptor expression and
the exhibited phenotype are caused by interactions with proteins which modify
the activity profile of the GPCR, such as activity modifying proteins (RAMPs) (Christopoulos A, 2003;
Fischer JA, 2002; Morfis M, 2003; Sexton PM, 2001; Tilakaratne
N, 2000; Udawela M, 2004). These protein functions
and interactions in different cells and cell types which we do not take into
account in our subcellular models pop up rather
unpleasant in the drug development process.
The popular techniques to
explore and analyze low-dimensional data at high speed are based on the idea
that this would provide all the data with sufficient predictive power to allow
for a bottom-up approach to drug discovery. The current High Throughput
Screening (HTS) and other early stage methods allow gathering low-dimensional
data at high speed and volume, but their predictive power is too low as they
lack depth of descriptive power (Perlin MW, 2002; Entzeroth
M, 2003). We are just clogging the drug development pipeline with
under-correlating data in relation to clinical reality. A bigger flow of
unmanageable data does not equal a higher correlation to clinical reality.
The knowledge gathered at
the infra-cellular level has to be viewed in its relation to the (living) cell
in its native environment and the biological and non-biological processes influencing its function and
health, which requires a top-down functional and phenotypical approach rather
than a bottom-up descriptive approach. Complex disease processes cannot be
explained by simple oligo-parametric low-level models. A high-speed
oligo-parametric disease model does not equal high predictive power. It is not
the ability to study a simplified disease model at high speed which will allow
us to succeed, but we must study and verify the functional outcome of the
in-vivo disease process itself.
A game of chess is not
described by naming its pieces, but by the spatial and temporal interaction of
both players or in other words the flow of actions and reactions, described in
a space-time continuum and if we add the color it is a spatio-spectro-temporal
flow of events. The individual pieces or moves do not explain the final outcome
of the game, only when the entire process is analyzed from a positional and
functional point of view we can understand and predict the reason why one
player wins or loses. You have to study a game of chess at the appropriate
organizational level in order to understand it or you will fail to find an
explanation for the outcome of the game.
Isocellular disease models
Cellular disease models
have already allowed us to study disease processes in geat detail, but they need to be
dealth with carefully. Validation of a cellular model and a thorough understanding of its
strengths and weaknesses is required. Using cellular disease models
in more detail is not a trivial endeavor. Cellular disease models need to be
related to at least the in vivo cellular disease process we want to study, so a
validation of this correlation is very important (Gattei V, 1993; Thornhill MH,
1993; Lidington EA, 1999; Dimitrova D. S., 2002).
We now know that metabolic
pathways show complex interactions and that gross genetic rearrangements can
impair entire parts of cellular metabolism. The cellular models used in
research should be validated for their functional and phenotypical
representation of in vivo, in-organism processes. However many popular cell
lines are not selected for their close linkage to clinical reality, but for
their maintainability in the laboratory, lack of phenotypical variation, ease
of transfectability, etc. . It is assumed that those cellular models are a
valid representative of the disease process, but almost never a thorough
assessment is being done. The phenotypic background of a cell has an important
impact on the structure and function of cellular proteins (Tilakaratne
N, 2000; Kenakin, 2003).
Primary cell lines cells in
general require a more complex tissue culture medium than most popular cell
lines. Cancer cells (and transformed cells) can usually grow on much simpler
culture medium. Replicative senescence and varying behavior at each passage
(which may necessitate a change of cell lines for long term experiments) also
make primary cell lines less popular, as they necessitate a change of cell
lines and variability in experimental data. Reduction of unpleasant variability
in experiments by choosing a specific disease model may create nice results,
but of a reduced predictive value. Quite often results obtained with one cell
line, cannot be confirmed by using another cell line, without even talking
about primary cells (Kenakin T, 2003).
CHO cells (Chinese Hamster
Ovary, Cricetulus griseus) are used in many assays, but they are not derived
from a human cell and are aneuploid (Tjio, J. H., 1958). HeLa cells are derived from an aggressive cervical
cancer; they have been transformed by human papillomavirus 18 (HPV18) and have
different properties from normal cervical cells (Gey, G.O., 1952). The U-2 OS
osteosarcoma cell line is easy to maintain and transfect (Ponten J, 1967). The
PC12 cell line which responds reversibly to nerve growth factor (NGF) has been
established from a rat adrenal pheochromocytoma, it has a homogeneous and
near-diploid chromosome number of 40 (Greene LA, 1967). HEC cells are derived
of a human endometrial adenocarcinoma cell line and are also very popular
(Kuramoto H., 1972). Two cell lines are very popular for epithelial barrier
studies: Caco-2 cells and Madin Darby canine kidney (MDCK) cells.
Caco-2 cells are the most popular cellular model in studies
on passage and transport, they were derived from a human colorectal adenocarcinoma
(Kirkland SC, 1986; Hidalgo IJ, 1989). Caco-2 cells are being used as a model to evaluate small intestine transport.
The interpretation of Caco-2 transport data is often confusing,
and is not always in agreement with in vivo observations, even when P-glycoprotein
(P-gp) is blocked by specific inhibitors (Hu, M., 1999; Hunter, J., 1993; Lennernas H, 1994).
Heterogeneity in the Caco -2 cell line, depending on passage number and origin of the cells,
leads to differences in transepithelial transport (Walter E., 1996).
Madin Darby canine kidney (MDCK) cells were isolated from a dog kidney (Gaush, C.R., 1966).
They are currently used to study the regulation of cell growth, drug metabolism, toxicity
and transport at the distal renal tubule epithelial level. MDCK cells are also used as
a cellular barrier model for assessing intestinal epithelial drug transport (Cho, M.J., 1989).
We quite often use the models which will grow in vitro, just to have at least
something, although we know that this is a highly uncertain approach.
Availability of a particular cellular model system does not equal predictability of the system.
Some popular cell lines may
correlate with themselves and not with the complex dynamics of the physiological
process in man they are supposed to represent. Studying the dynamics of the
involvement of a protein in a disease in patients and transforming this
knowledge into a disease model in a particular cell line requires a careful
assessment before embarking on a drug discovery process. Functional cell model
drift should be verified at regular intervals and taken into account.
Even within individual cell
lines there is not always homogeneity in phenotype and function. Cancer cells
show genetical and chromosomal instability as they tend to lose parts of
chromosomes (Duesberg
P., 1998; Lengauer C, 1998; Duesberg P, 2004).
Using cell lines derived from cancers poses a correlation risk in relation to
clinical reality on research done by using these types of cell lines.
Continuous sub-cultivation of cells and an increase in the number of passages
may lead to chromosome rearrangements and loss of functional reactivity
(Dzhambazov B, 2003). Loss of function destabilises a cell when critical parts
of pathways are lost, although cell cycling may continue in parts of the cell
culture, but this will cause a drift and on experimental results.
Many of the most popular
cell lines lack parts or even entire chromosomes and therefore large chunks of
metabolic pathways. A drug molecule can not interact with the proteins which
are not present in the cell line and an adverse or even positive effect will go
unnoticed. Functional
loss of proteins and enzymes in cancer cell makes them unresponsive to drugs if
the protein(s) which are the target of a drug are lost without killing the cell
as such.
Even when a protein is successfully
expressed in a cell as shown on a Western blot, this does not equal functional
success. Western blotting tells you how much protein has accumulated in cells.
Even knowing the rate of synthesis of a protein by Radio-Immune Precipitation
(RIP) does not predict the functional outcome of protein expression. Protein
function is also depending on the metabolic background of the cell in which the
protein is expressed and its spatial and temporal organisation. If the
enzymatic and structural background of the cell does not meet the prerequisites
to put a functional protein in the right location, embedded in the right
functional environment, nothing appropriate will happen. An appropriate
functional assay is required to validate proper function of the expressed
protein.
Conclusions drawn from
phenotypically uniform and simplified cell lines do not show a reliable or
constant correlation to in-organism cellular dynamics. A functional
comparison between isolated native cardiac myocytes and cloned hERG demonstrates
the advantages of cardiac myocytes over heterologously expressed hERG channels
in predicting QT interval prolongation and TdP in man (Davie C, 2004). The
involvement of heterotrimeric G proteins in cell
division was only discovered by looking at the native protein in its natural
environment and not by using a trasfected system
where the physiological gene regulation is disabled (Zwaal RR, 1996; Kimple RJ, 2002).
The location of
glycosyltransferases involved in N- and O-glycan chain elongation was traditionally
found to be confined to the Golgi-apparatus in phenotypically simple cells,
such as HeLa cells. However, localization studies conducted in primary cell
cultures often reveal ectopic localizations of glycosyltransferases usually at
post-Golgi sites, including the plasma membrane (Berger EG., 2002). This shows
that the prototypical or average cell does not exist in drug discovery as a
valid broadrange cellular model. Cells are not just
interchangeable containers with molecules in which we can pour reagents but
highly dynamic environments.
In vivo enzymatic reactions
are not linearly correlated to protein concentration or of zero order. The
intracellular environment causes a more complex functional pattern for a given
protein, such as bell shaped relation between protein concentration and
function. A blunt on/off expression in a transfected cell does not correlate
well to the physiological condition in a primary cell. When the appropriate
metabolic environment is not present when studying a protein in a cellular
disease model, predictivity of the disease model may be low compared to
physiological conditions.
A traditional (homogeneous)
cell culture in the laboratory may not yet mimic the physiological conditions
in an entire organism, so our approach to cell-based research (and beyond)
requires some redesign also. Creating a virtual organism, by differential
screening of a multitude of cell type representing the main cell types in the
human body (cardiomyocytes, hepatocytes ) could help us to improve the
predictive value of cellular disease models. We need to study cell-to-cell and
cell-type-specific pathway dynamics in more detail, as is the case for nuclear
factor-kappaB (NF-KappaB) (Schooley K, 2003). Studying Biologically Multiplexed
Activity Profiles (BioMAP) directly in primary cells provides us with results
which are closer to the situation in man. BioMAP profiling can allow
integration of meaningful human biology into drug development programs (Kunkel
EJ, 2004).
Metabolic pathways in cells
do not exist in a void, but are interconnected and highly dynamic processes.
Blocking a pathway has far-reaching consequences for the intracellular
environment. The upstream metabolites will either find their way through other
metabolic pathways or pile-up. Some inborn errors of metabolism are an example
of this principle (PKU ). Drugs blocking pathways also cause a distortion of
the delicate balance in metabolic processes and may cause upstream effects by
metabolites which are normally metabolized before they can cause any harm. The
kinetics of the pharmakon may be documented, but the change in cellular
metabolism and pathway-network distortion are less well understood. Upstream
metabolites may become processed by other pathways and unexpected adverse effects
may show up. Adverse effects on cellular metabolism are only present in those
cells which have an intact metabolic pathway and not even all cell types
activate the same pathways at all times.
Cell cycling and circadian
oscillators are a source of periodic variation in our cell based experiments
(Okamura H., 2004). Oscillations of cellular functions are to be explored and
taken into account, as they act as a dynamic background or reference level
representing a dynamic reference for studying cellular structure and function.
Oscillations which are not accounted for act as periodic noise, disturbing our
measurements and masking subtle, but possibly important events in our
experiments.
Differential multiplexing
in (high-content) cell based screening could help us to acquire more
information about the spatial and temporal dynamics of cellular processes.
Developments are going on towards experimental multiplexing and up-scaling of
the capacity of quantitative cellular research (Perlman ZE, 2004). Techniques such
as High-Content Screening (HCS) or multiplexed quantification can be applied to
cellular systems to study intra-cellular events on a large scale (Van Osta P.,
2000; Taylor DL, 2001; Van Osta P., 2002b; Abraham VC, 2004; Van Osta P.,
2004). Subcellular differential phenotyping is
already possible on a large scale by using human cell arrays. We can use
multiplexed molecular profiling in cells to obtain more information (Stoughton
RB, 2005). Light-microscope technology is used to explore the spatial and temporal
dynamics in cell arrays in great detail (Ziauddin J, 2001; Bailey SN, 2002;
Baghdoyan S, 2004; Conrad
C, 2004; Hartman JL 4th, 2004).
Analyzing a large number of
tissues for candidate gene expression is now greatly facilitated by using
Tissue MicroArray (TMA) technology (Kononen J, 1998; Simon R, 2002;
Braunschweig T, 2004).
From individual cell to cytome
Studying cell function and
drug impact at the level of the individual cell is called cellomics (Russo E.,
2000). However, the concept of cellomics does not take into account the
supra-cellular heterogeneity which is present in every cellular system, such as
a cell culture or an organism. By studying cells while ignoring their diversity
we make the same mistake as the statistician who drowned crossing a river that
on average was just three feet deep.
Due to the heterogeneity of
cell types and differences between cells in a healthy and disease state, we
need to take this heterogeneity into account. Cytomes can be defined as
cellular systems and the subsystems and functional components of the body.
Cytomics is the study of the heterogeneity of cytomes or more precisely the
study of molecular single cell phenotypes resulting from genotype and exposure
in combination with exhaustive bioinformatics knowledge extraction (Davies E,
2001; Ecker RC, 2004b;
Valet G, 2003; Valet G, 2004).
In order to get the broader
view on pathological processes, we should move on to the phenotypical and
functional study of the cellular level or the cytome in order to understand what is
really going on in important disease processes. Although the genome and
proteome level have their predictive value in order to understand the processes
involved in disease (and health), the cytome level allows for an understanding
of pathological phenotypes at a higher level. By integrating the knowledge from
the genome and proteome, we could give guidance to the exploration of the
cytome, which was not possible before this knowledge was available.
The cytome level will also
provide guidance to focus the research at the genome and proteome level and so
creating a better cross-level understanding of what is going on in cells (Gong
JP, 2003; Valet G, 2004; Valet G, 2004b). Some would see this as taking a step
back from the current structural and systematic descriptive approach, but it is
mainly a matter of integrating research at another level of biological
integration and looking in a different way to the web of interactions going on
at the cellular level. Biological processes do not exist in a void, but they
are a part of a web of interactions in space and time, rather than being an
island on their own. A cell is a multidimensional physical structure (3D and
time) with a finite size, not a dimensionless quantity. We cannot ignore the
spatial and temporal distribution of events, without losing too much
information.
In recent years the tools
have matured to start studying the cellular level of biological integration,
but the tools are still used in the same way as if they were derived from
low-content high-throughput phenomena as this is still the dominant research
model. The tools to generate and explore a high-dimensional feature space are
still scattered and not brought into line with the exploration of the cytome.
Functional processing in cellular pathways
The interconnection of
genome, proteome and cytome data will be necessary in order to allow for an
in-depth understanding of the processes and pathways interacting at the
cellular level. A monocausal approach will have to be replaced with a poly- and
pluricausal approach in order to understand and explain the phenomena going on
at the cellular level. Pluricausal means causal contributions at different
levels, such as genes, other cells and environmental influences. Polycausal
means multiple causal contributions at the same biological level, such as
polygenic diseases or multiple agonistic and antagonistic environmental
influences. The concept of a multithreaded, multidimensional, weighed causality
is needed in order to study the web of interactions at the cellular level. A
drug modulates cellular function, but changes can be studied at different
levels of biological integration:
Disease outcome = drug x (a
x clinicaln
+ b x physiologicalp
+ c x cellularq
+ d x geneticr
)
Disease models should incorporate
mixed and nonlinear effects. Diagnosis and drug
discovery merge if we take parallel models for both. The clinical diagnosis or para-clinical diagnosis of a disease should show a high
correlation with the disease models used to study its possible treatment. A
cause (e.g. a single gene defect, a bacteria) can have multiple consequences
and as such be poly-consequential, which is the mirror situation of a single
consequence being caused by multiple causes (co-causality or co-modulation)
acting either synergistic or antagonistic (e.g. a disease with both a genetic
an environmental component). In reality, a pathological condition is a mixture
of those extremes (e.g. a bacterial or viral infection and the hosts immune
system) and as such a simple approach is not likely to succeed in unraveling
the mechanism of a disease. With the current systematic and descriptive
approach however, we get lost in the maze of molecular interactions. We are
looking at too low a level of biological integration and we get lost in a maze
of structures and interactions. The cell is the lowest acceptable target, not
its single components, like DNA or proteins.
We are looking at the
alphabet, not even words or sentences, nature is not a dictionary, but it is a
novel. We should study the flow of events in a cell with more power, not only
the building blocks. As an example, Mendel did not need to know about DNA in
order to formulate his laws of inheritance and he did not know that the
discovery of the physical carrier of inheritance, DNA, would confirm his views
later on, but his laws are still valid as such. Certainly physics was not at
the stage it was in the 20th century when
The value of a scientific
model does not lie in the scale of phenomena it describes, but in its
predictive correlation to the reality it tries to capture. The more we may try
to exclude elements from reality, the better we may be able to build a model
which holds in a tightly controlled situation in our laboratory, but fails when
challenged by full-blown reality in the outside world.
What we find should not be
in contradiction to what lower level structural descriptive research discovers,
but we should not wait for its completion to start working on the problems we
are facing in medicine and health care today.
Epicellular disease models
Epicellular models are of great importance for both
efficacy testing as well as for toxicity testing. Organoids, parts of organs,
isolated organs and animals are being used as epicellular disease models.
The advantages of in vitro systems in toxicity
testing are numerous. In vitro tests are usually quicker and less expensive. Experimental conditions
can be highly controlled and the results are easily quantified. However, the relative simplicity of
nonwhole-animal testing results in limitations as well. Cells or tissues in culture cannot predict
the effect of a toxin on a living organism with its complex interaction of nervous, endocrine,
immune, and hematopoietic systems. In vitro systems can predict the cellular and molecular
effects of a drug or toxin, but only a human or animal can exhibit the complex physiological
response of the whole organism, including signs and symptoms of injury.
Rats, mice, dogs, (non-human) primates, rabbits, cats, guinea pigs, hamsters, miniature swine,
goats, farm pigs, etc. are some of the species which are available for preclinical and non-clinical evaluation.
The main reasons for wanting to predict human sensitivity are that most failures in the
clinic are due to safety problems and lack of effectiveness. The inability of animal
models to predict these failures before human testing or early in clinical trials
dramatically escalates costs. The choice of the relevant animal species for preclinical
studies is an important consideration. The goal of pre-clinical safety assessment
studies is ultimately an estimate of safety in humans. A critical assumption is
that the toxicity responses seen in various animal models will be reflective of
those in humans (e.g. comparative toxicogenomics). Assumptions require in-depth validation,
some of which we will discuss in this section.
Animal models are an important part of the drug discovery an development process and they have
made a significant contribution to our understanding of disease processes. The correlation of
the animal model to the actual process (efficacy, toxicity) in man is an important issue to
consider (Hondeghem LM, 2002; Huskey SE, 2003). Inter-species differences, can have a significant impact
on the interpretation of results in an animal model for a human disease. Earlier
successes do not relieve us from continuing critical assessment of every model.
With the increasing complexity of the diseases being studied, subtle interspecies differences
become increasingly important. In the past twenty years a lot has changed in the use of
animal models to study human disease processes and develop new drugs.
A first example of using an epicellular screening model for drug discovery was the discovery of
Arsphenamine (Salvarsan) by Paul Ehrlich. Paul Ehrlich (1854-1915) started the first screen in search of
compounds effective against syphilis, resulting in the world's first "blockbuster" drug Salvarsan.
Sahachiro Hata discovered the anti-syphilitic activity of this compound in 1908 in the laboratory of Paul Ehrlich,
during a survey of hundreds (606) of newly-synthesized organic arsenical compounds.
This was the first organized team effort to optimize the biological activity of a lead compound through
systematic chemical modifications, the basis for nearly all modern pharmaceutical research.
What was different from modern-day High-Throughput Screening (HTS) is that their mouse model was not only a
valid representation of the disease Syphilis in man, but also in one pass provided information about ADME and
Toxicity for which their mouse model provided sufficient predictive power. Essentially we now perform screening
in test-tube enabled models, even if we now call them multiwell-plates.
Only disease processes which can be represented in a test-tube or agar-plate are amenable to ultra-high automation,
but in many cases they are a "reductio ad absurdum" of the full blown human disease process
(the premise that the test-tube assay represents the disease with enough validity to start from here is often false).
The validity of simple-model screenings to represent the complexity of a disease process in man is of secondary
importance as we hope to compensate for this later on in the process (we do, but at an unacceptable cost).
Another historical example of a drug discovered by using an animal model for a disease, is the discovery
of the first successful oral antibiotic, Prontosil (Sulfamidochrysoidine). Prontosil was developed by
Gerhard Domagk, working in the Bayer sector of
I. G. Farbenindustrie. Sulfamidochrysoidine seemed to have no effect on bacteria in vitro, but Domagk
went ahead and tested it on 26 mice injected with streptococci. Fourteen were kept as controls,
and 12 were treated with Prontosil. All of the controls died within a few days, while all of the
treated mice survived. Later Gerard Domagk received the
Nobel Price
for his discovery, which saved the lives of many people.
Much has changed since Gerard Domagk's discovery of Prontosil. Animal models are being used for
evaluation of efficacy and toxicity. We can now use genetically
modified animals to study gene regulation and cell differentiation in a
mammalian system (Gordon JW, 1980; Isola LM, 1991; Brusa R., 1999). Transgenic
and gene-deleted (knockout) mice are used extensively in drug discovery
(Rudmann DG, 1999). The pioneering work of Mario Capecchi, Martin Evans, Oliver Smithies and others
has enabled the construction of increasingly sofisticated animal models (Smithies O, 1984, Evans MJ., 1989).
Homologous recombination between DNA sequences residing in the chromosome and newly introduced,
cloned DNA sequences (gene targeting) allows the transfer of any modification of the cloned gene
into the genome of a living cell (Capecchi MR., 1989). The challenges now are to model the complex
multifactorial diseases, instead of simple monogenic diseases (Smithies O., 1993; Smithies O., 2005).
Simple knockouts are usually designed to lead to loss of protein function, whereas a subset of
cancer-causing mutations clearly results in gain of function. Mored dynamic transgenic mouse
systems are now avaialable (Sauer B., 1998; Maddison K, 2005). The original Cre and FLP recombinases
have demonstrated their utility in developing conditional gene targeting, and now other
analogous recombinases are also ready to be used, in the same way or in combined strategies,
to achieve more sophisticated experimental schemes for addressing complex
biological questions (Garcia-Otin AL, 2006).
Although knockout technology is highly advantageous for both biomedical research and drug development,
it also contains a number of limitations. For example, because of developmental defects, many knockout
mice die while they are still embryos before the researcher has a chance to use the model for
experimentation. Even if a mouse survives, several mouse models have somewhat different physical
and physiological (or phenotypic) traits than their human counterparts. An example of this phenomenon
is the p53 knockout. Gene p53 has been implicated in as many as half of all human cancers. However
p53 knockout mice develop a completely different range of tumors than do humans. In particular,
mice develop lymphomas and sarcomas, whereas humans tend to develop epithelial cell-derived
cancers. Because such differences exist it cannot be assumed that a particular gene will exhibit
identical function in both mouse and human, and thus limits the utility of knockout mice as
models of human disease (Pray L., 2002).
There is not only interspecies variation, but also
intraspecies variation. The biological variability is also present in genetically modified animals.
We expect a specific phenotype from a specific genetical modification.
In genetically modified mice however, the observed phenotype is not always the direct result of the
genetic alteration (Linder CC., 2001; Schulhof J, 2001). The effect of the
genetic modification is not completely straightforward, due to variations in
the genetic background of the animals (Crusio WE., 2004). Transgenic mice
containing the same genetic manipulation exhibit profoundly different
phenotypes due to diverse genetic backgrounds (Sigmund CD., 2000; Sanford LP,
2001; Holmes A, 2003; Thyagarajan T, 2003; Bothe GW, 2004).
The relevance of
a particular model is linked to the similarity of a process in the model animal and man. There is however
considerable variation between species which complicates the use and evaluation of animal
models (Hucker HB., 1970; Smith CC., 1967; Smith RL., 1974; Hengstler JG, 1999; Chiu SH, 1998; Nelson SD., 1982;
Fry JR., 1982). A required part of drug development is the "Chronic Bioassay". Hundreds of pharmaceuticals
have been reported to give a positive result in the standard "Chronic Bioassay", which
consists of an 18 to 24 month daily administration of the test compound in mice and rats. This is
in contrast with 20 pharmaceuticals, which are known to be carcinogenic to humans (Van Deun K, 1997).
Interspecies differences are an important issue and require in-depth validation (e.g. comparative toxicogenomics).
Animals are popular models for studying G.I. absorption for oral drug uptake, but in addition to metabolic
differences, the anatomical, physiological, and biochemical differences in
the gastrointestinal (G.I.) tract of the human and common laboratory animals can cause significant
variation in drug absorption from the oral route (Kararli TT., 1995).
A classical example of inter- and intraspecies variability is Thalidomide. The mouse and rat were resistant,
the rabbit and hamster variably responded, and certain strains of primates were sensitive to thalidomide
developmental toxicity. Different strains of the same species of animals were also found to have
variable sensitivity to thalidomide (Neubert D, 1988; Bila V, 1989).
Although the drug was marketed in 1957, reproductive studies on thalidomide in animals were not started until
1961, after the drug's effects on human fetuses had begun to be suspected (MacBride, 1961).
Initial studies on rats and mice revealed some reproductive abnormalities, notably reduction in litter size due
to resorption of fetuses; however, only when the compound was tested in the New Zealand white rabbit did
abnormalities similar to those noticed in human babies occur (Cozens DD., 1965). Studies on monkeys revealed that they were
almost as sensitive as humans to the deformative effects of the drug (Delahunt CS, 1965).
Mouse models require careful evaluation and validation. Selection of mouse models of cancer is often based simply on availability of a mouse strain and a known compatible tumor. As a consequence cancer models in mice quite often fail to predict success in clinical development later on (Kelland LR., 2004; Kerbel RS., 2003; Peterson JK, 2004; Schuh JC., 2004; Voskoglou-Nomikos T, 2003).
Using inbred mouse strains
reduces variation in genetic background, but also reduces the correlation of
the disease model to real-world genetic and metabolic variation encountered in
human populations. Finding a strong correlation in an inbred laboratory animal
population is no guarantee that the correlation will hold in an out bred
natural population (Mackay TF., 1996; Macdonald SJ, 2004). Do we want nice
results with a low standard deviation (SD), or do we need results highly
correlating with clinical reality? If
one wishes to obtain the optimal mouse model for a human disease, one needs to
choose the correct genetic background as well as the correct mutation (Erickson
RP., 1996).
Careful validation of a transgenic mouse model is required, such as with models for
Alzheimer's Disease, for example by using biomarkers and PET (Bacskai BJ, 2003;
Klunk WE, 2004; Klunk WE, 2005). Validation of a model for a complex disease
requires a thorough validation.
We still do not have an
in-depth understanding of the delicate spatial and temporal interplay in
metabolic pathways in cells, organs or entire organisms in transgenic animals.
Introducing or removing a gene without a clear understanding of its spatial and
temporal expression pattern, leaves us with a correlation deficit in relation
to the disease process in man.
When we modify a gene, we
modify a pathway-web with upstream and downstream consequences for cellular
metabolism in different cellular compartments (nucleus, Golgi ). The
metabolites which (dis-) appear due to the modification will modify a highly
dynamic network of metabolic interactions. In-vivo spatial and temporal
variation in protein structure and activity profiles will add to the complexity
of unravelling the functional impact of modified gene expression.
When we want to bridge the gap from "model to man" we must identify and verify the
process in its native environment the cell and the entire organism (e.g. with a biomarker)
and compare and validate what happens in a mouse to what happens in man (Lee JW, 2006). The use of biomarkers
can be added to preclinical studies to help justify the choice of animal species.
The ultimate animal model for compound selection is still man himself and with the
recent introduction of extremely sensitive detection methods such as accelerator
mass spectrometry (AMS) and Positron Emission Tomography (PET) it is now possible
to evaluate the absorption, metabolism and elimination of compounds in man as
part of the pre-clinical selection process (e.g. microdosing).
Scientific evolution and when Biology meets Chemistry
Before we enter the road towards the development of a new drug we still lack a lot of knowledge and understanding of the
complex biology of the disease pathway. The long way towards the application of a new drug or biological for a human disease
starts nowadays at the lowest possible level of biological complexity; the confrontation of a single molecule with a single molecule.
We remain blind for the complexity of the intrahuman ecosystem until very late into the development cycle of new medication
which is why there is a bias towards false positives built into the process. At the moment we just accept this and we still succeed
against all odds to bring new medication to the market. The overall system works, but is not an example of an efficient and effective
process, we just accepted to live with these low odds of success.
The fundamental mechanism by which science progresses is the same as how nature evolves by means of natural selection.
In science the role of mutations is performed by new paradigms which break through the frontiers of established science and provide
a comptitive edge though their improved ability to explain reality. However more than 99 percent of all experimetns are performed within
the safe environment of present day scientific principles. Science evolves by means of scientific selection, which is somewhat different
from natural selection as the selection pressure is
partly exercised by man in an artificial laboratory environment instead of nature itself (e.g. peer review, commercial pressure,
present day scientific theory and paradigms.
The scientific selection process therefore tends to produce a bias towards false positives, which becomes clear at the end of the process
when confrontation and selection by nature itself can no longer be avoided. This happens when we have to face clinical reality
itself and not only the artificial environment of the laboratory. The quality of the selection pressure in the end
determines the potential speed and direction of scientific evolution.
Figure 32. Biology and Chemistry meet at the point of minimal system complexity. The bandwidth of system selection pressure is minimal at the moment of confrontation. |
Figure 33. Proportion of understanding achievable within chemical and biological system space. Biology is more complex than molecular chemistry, we do not master each one completely. |
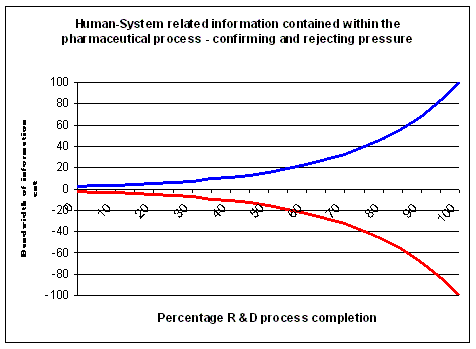
Figure 34. Evolution of system selection pressure during the R&D process. Selection pressure evolves in a non-linear way. |
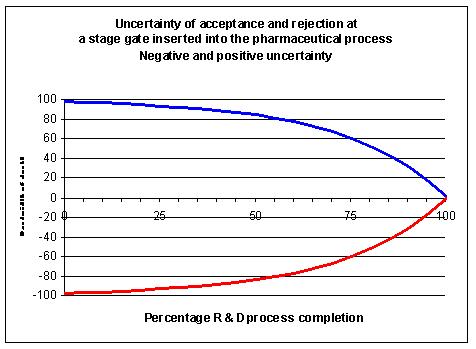
Figure 35. Bandwidth of uncertainty during the R&D process. What is not present at the moment of decision, does not contribute to the decision. |
Working with models at different levels of biological complexity, has its consequences.
We arrive at the point of confrontation between biology and chemistry at a moment when we have
reduced system complexity to its bare minimum (Figure 32). The long an winding
road towards unraveling the disese process and arriving at a screenable target has made us strip away
man from the target molecule. Almost nothing is left of the intrahuman ecosystem at the moment when
we confront chemistry with biology. Less than 0.01 % of the full blown selection pressure is
present at the moment of confrontation between biology and chemistry. We are as such 99.99 % blind
for interfering phenomena. The molecule surviving the initial selection process evolves in an almost
pristine environment compared to the metabolic jungle of man. Unable to increase our understanding
of biological processe in the intact human ecosystem, fast enough to keep productivity of the R&D process sustainable,
we retreated into simplified models in the hope to catch up later on in the process.
When we slowly retreat back to the confrontation with the full blown intrahuman ecosystem, we make our decisons
at each stage gate within an environment which is only partly representative for the ultimate situation (Figure 33).
Instead of symplifying at each step (as is the case during unraveling a disease, left side of Figure 32), we are confronted
with a system of higher complexity after passing a stage gate, for which the previous step provides less than ideal predictive
power. Even if we extract all information before arriving at a stage gate, our predictive power is flawed in relation
to increased complexity awaiting us after the gate. We see what we observe, but not what we need to know.
The retreat is also not following the same trajectory as we used for approaching the biological-chemical confrontation, but a
twisted path biased by regulations and interspaced with gaps, caused by model discontinuities.
The relative power of our understanding in relation to the system we are working in, is higher in the early
chemical environment than in the later biological environment (Figure 34). As we can witness
in the high late stage attrition rates, the overall chance of succes of Phase III now equals about 50 %, or it is as
predictive as tossing a coin.
The selection pressure, both positive and negative reaches a high level only at the end of the pharmaceutical R&D process,
because it is only then the full blown complexity of man and human population is able to exert its effect (Figure 34).
The consequence is that the power of a decision at a stage gate is highly uncertain during most of the process
(Figure 35). Increasing the capacity of under-predicting processes early on in the pipeline, will not bring down
attrition rates.
Less questions, better answers
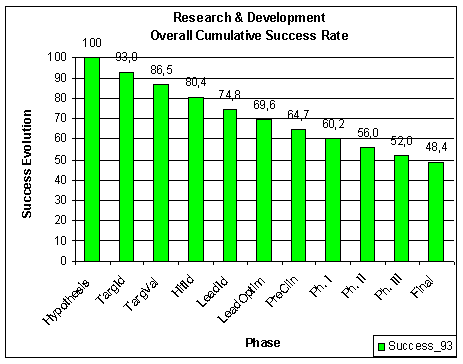
Figure 36. A drug discovery and development process with a right choice of 93% at each step still only performs at about 50% overall. By Phase III we spend 50% of our efforts on failures. |
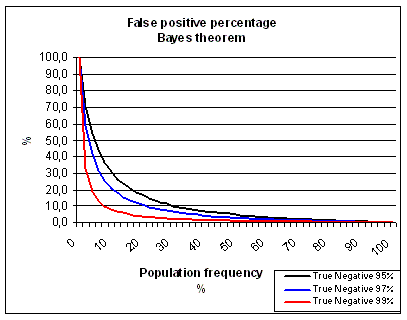
Figure 37. Searching for needles in a haystack leads to very high false positive rates. |
The overall drug discovery and development process is Data Rich, but Information Poor (DRIP),
which explains its low overall efficiency. Within major pharmaceutical companies, there are
numerous functional silos generating different data from different technologies with different levels of error.
A process as complex as the present-day drug discovery and development process requires
a success rate (true positives and true negatives) of more than 93% at each step to perform
above 50% overall (Figure 36). Even if we make the right choice in 93% of all
cases at each stage-gate and make a false positive or false negative choice in only 7%, this means that by the end
we will spend 50% of our efforts on failures in Phase III. The wrong choices we make in the beginning we take with us
through the entire process. Every stage succeeds or fails at its own stage-gate, but the overall process fails
because the decisions at each stage-gate are under-correlating to the true end-point of the process
(cure a disease in man, not in a test-tube). We confirm or reject the past (up to the stage-gate), but do not
(enough) predict the future state of the process and
so we add-up correlation deficits (false positives) with each step. At the moment the choice of target and lead candidate is flawed, due to a
lack of knowledge about the disease process and the interaction of the chemical substance with the human biological
system. We fail at the beginning, but pay the price at the end. As we were incapable to understand molecular processes in
complex biosystems, we added some simplified, but high-speed environments upfront to the process. We increased quantity, but in
the process sacrificed the quality of correlation with the reality of disease in man.
False positives are a problem in any kind of test: no test is perfect, and every the test will incorrectly report a
positive result in certain cases. The problem of false positives lies, however, not just in the chance of a false
positive prior to testing, but determining the chance that a positive result is in fact a false positive.
Using Bayes' theorem, if a condition is rare and only a minority of molecules show activity in a
High Throughput Screening (HTS) campaign, then the majority of positive results will be false positives, even if the assay for that
condition is (otherwise) reasonably accurate (Figure 37).
Blind screening of a random chemical library (e.g. generated by combinatorial chemistry) against a biological target
leads to an excessive number of false positives, putting more pressure on the secondary screens and beyond to weed out
these false positives. A simple example, based on
Bayesian inference illustrates the phenomenon.
An assay is being used which has a 99.99% true positive rate. The graph shows the percentage
of false positives for three different true negative rates 95%, 97% and 99%. As you can calculate yourself
(see Bayesian inference), the true negative rate has a more
dramatic impact on the number of false positives when the population frequency (hit rate) is very low.
By using a chemical library whose chemical space is badly matched to the biological space of the target we create an
high proportion of false positive results. As the high attrition rates in clinical development show (e.g. Phaze II)
show, we do not get rid of these false positives until very late in the overall process.
However when the population frequency of chemical structures matching
with the validated target increases, the rate of false positives decreases exponentialy (Figure 37). Designing
molecules with an understanding of their potential interaction with a truly validated biological target leads to
an increase in process efficiency which surpasses all other attempts made to improve the drug discovery and
development process. Mistakes made at the beginning are harder to compensate for later on in the process. It is
at the start of the process when the path towards a succesful drug in man heads in the right direction or not. We first need
to understand what happens in the cytome of man, not only in an Eppendorf. Now the tools are becoming available to achieve this goal:
The idea of a Human Cytome Project is about the improving ability to understand system-wide phenomena
at a (sub-)cellular resolution (molecular physiology). This leads to being able to ask the question about effectiveness and
toxicity in one step in a complex biological environment, in model organisms and in man (ask less questions,
but get better answers). Molecular observation and answers to system-wide questions leading to better models generated from
improved observations.
Human Cytome Project - the way to go
Figure 38: Exploring the organizational levels of human biology for the benefit of medicine.
A linked and overlapping cascade of exploratory systems
each exploring -omes at different organizational levels of biological systems
in the end allows for creating an interconnected knowledge architecture of entire cytomes.
The approach leads to the creation of an Organism Architecture (OA)
in order to capture the multi-level dynamics of an organism.
Practical issues on how to to explore the human cytome and a concept for a
software architecture can be found on these pages:
The article "How to explore and find new directions for research",
discusses ways to explore the cytome, ranging from digital microscopy, High Content Screening (HCS) to Molecular Imaging.
The cytomic microcosm, both in-vivo ans well as in-vitro is accessible for highly detailed detection and analysis.
New experimental techniques allow for an in-depth exploration of the (patho-)physiology of the human cyctome. The
human cytome is now more accessible than ever for exploration and as a consequence a leap forward in our understanding
of the complex machinery of the cytome in health and disease.
The article "A framework concept to explore the human cytome on a large scale",
presents some concepts on the way towards large scale exploration of the human cytome system.
Final remarks
The war against human diseases is not an easy one to win. It spans the globe and stretches through the ages. The trenches of our war against disease reach from bench to bedside and require a strong and united effort in order to succeed.
Innovation is much more than discovery of new drug targets. There is no substitute for knowledge and understanding.
More rational, more efficient and more informative preclinical and clinical drug development is required. We have to get away from
a trial and error approach to a "cognitive" chemical biology approach, matching biological and chemical space.
We have to do a better effort to build up understanding about the target (system) and active compounds.
Drug discovery and development show a decreasing efficiency in relation to the amount
of money being invested. The level of understanding at the end of discovery
(and preclinical development) should achieve a knowledge level which is capable
to predict success at the end of the pipeline much better than we do now. We need
to improve our preclinical disease models and our understanding of clinical reality. The quantity of what we can achieve with
automation of drug discovery needs to be matched with the predictive quality of the science
underlying the automated procedure. There is an urgent need to improve the productivity of
drug discovery and development, but this time we should evaluate the underlying process better than
we did with the advent of target-based drug discovery and High Throughput Screening (HTS). A high
statistical significance, does not guarantee a concommitant clinical relevance, as correlations
derived from inductive reasoning may be flawed in relation to the clinical reality they are meant to
represent within the experiment.
Our disease models should capture more of the high-order complexity of biology, beyond the genes and proteins which are the current focus of research. Higher-order disease models should place genetic and protein research results in a broader perspective and complement them with information about high-order interations in space and time. We should study cells with taking into account their in-cytome differentiation and their real-life behavior. The sooner NCEs or NBEs evolve in a "rich" or lifelike biological environment (background, population variability) the earlier we capture (un-)wanted phenomena. It is all about improving the Probability Of Success (POS) for the overall process. There is no escape from the demands for better treatments from patients, society and shareholders. Scientists are working day and night to develop new treatments for unmet medical needs, but their effort should become more efficient and effective, so more of the candidate drugs reach the patients waiting for new and improved treatments. The weight of higher-order biological exploration should increase in the overall discovery and development process. More of man's diseases should be captured in our (pre-)clinical disease models.
A possible way out is to start drug discovery at an intermediate level of biological complexity, the cellular level (the cytome, taking into acount physiological cellular heterogeneity), with analysis of phenotypical and functional parameters (Cellular Physiology or Phenotype Based Screening). Deconvolve back towards molecular targets, which now have been confronted with drug candidates in an already complex biological system.
In the end process improvement should lead to a dramatic decrease in false positive results in preclinical development, while at the same time avoiding an increase in false negatives. This should lead to a better performance of the overall drug discovery and development pipeline in which more and better drugs reach the patients. I want to end this article with a quote from Dr. Paul Janssen: "For many sick people, there are still no drugs, and it is our job to develop good medicines".
Acknowledgments
I am indebted, for their pioneering work on automated digital microscopy and High Content Screening (HCS) (1988-2001), to my former colleagues at Janssen Pharmaceutica (1997-2001 CE), such as Frans Cornelissen, Hugo Geerts, Jan-Mark Geusebroek and Roger Nuyens, Rony Nuydens, Luk Ver Donck, Johan Geysen and their colleagues.
Many thanks also to the pioneers of Nanovid microscopy at Janssen Pharmaceutica, Marc De Brabander, Jan De Mey, Hugo Geerts, Marc Moeremans, Rony Nuydens and their colleagues. I also want to thank all those scientists who have helped me with general information and articles.
References and background
References can be found
here
Some background on the idea can be found
here
Cytome Research
- Max-Planck-Institut für Biochemie
- Indiana Metabolomics and Cytomics Initiative (METACyt)
- Purdue University
- Bindley Bioscience Center at Discovery Park
- Carnegie Mellon University
- Coriell Institute
- Herzzentrum Leipzig GmbH
- Prince Felipe Centro De Investigation - Cytomics Laboratory
- Catholic Univ. Leuven - Functional cytomics for cell therapy
- Ehime University - Laboratory of Molecular Cell Biology
- Ehime University - Laboratory of Molecular Cell Biology - Section of Cytomics
Additional Information
- Towards a Human Cytome Project
- Draft: Human Cytome Project
- Biomedical Imaging
- Healthcare in Motion
- Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products
- Innovative Medicines Initiative (IMI)
- European Medicines Agency (EMEA) Road Map to 2010
- New Safe Medicines Faster Project
- Priority Medicines for Europe and the World Project "A Public Health Approach to Innovation"
- Moving Medical Innovations Forward? New Initiatives from HHS
- OECD -Innovation and Technology Policy
- Cytomics
- E-Cell Project
- Physiome Project
- EuroPhysiome - STEP
- IUPS Physiome Project
- GIOME Project
- Functional genomics
- Cyttron
- National Resource for Cell Analysis and Modeling - NRCAM
- Prediction in Cell-based Systems (Predictive Cytomics)
- Human Genome Project
- Post-Human Genome Project Progress & Resources
- How Many Genes Are in the Human Genome?
- NCBI
History of Human Cytome Project set of articles
Previously posted versions, sorted by date
Copyright notice and disclaimer
These webpages
represent my personal interests, opinions and ideas, not those of my employer
or anyone else. I have created these web pages without any commercial
goal, but solely out of personal and scientific interest. You may download,
display, print and copy, any material at this website, in unaltered form only,
for your personal use or for non-commercial use within your organization.
Should my web pages or portions of my web pages be used on any Internet or
World Wide Web page or informational presentation, that a link back to my website (and
where appropriate back to the source document) be established. I expect at
least a short notice by email when you copy my web pages, or part of it for
your own use.
Any information here is provided in good faith but no warranty can be made for
its accuracy. As this is a work in progress, it is still incomplete and even
inaccurate. Although care has been taken in preparing the information contained
in my web pages, I do not and cannot guarantee the accuracy thereof. Anyone
using the information does so at their own risk and shall be deemed to
indemnify me from any and all injury or damage arising from such use.
To the best of my knowledge, all graphics, text and other presentations not
created by me on my web pages are in the public domain and freely available
from various sources on the Internet or elsewhere and/or kindly provided by the
owner.
If you notice something incorrect or have any questions, send me an email.
This article was created by Peter Van Osta.
Email: pvosta at gmail dot com
A first draft was published
on
Latest revision on 10 May 2014